Генетика людини: методичка
5.4. Генні хвороби
Сьогодні відомо близько 4500 генних захворювань, характер успадкування яких визначається законами Г. Менделя. (Див. розділ 4.1.). Вони складають численну та різноманітну за клінічною картиною групу патологій, в основі яких лежить мутація одного гена.
Середня загальна частота новонароджених з генними хворобами складає 1%. Із них 42% уражені аутосомно-домінантними патологіями, стільки ж – аутосомно-рецесивними, 15% – зчепленими з Х-хромосомою та 1% – Yзчепленими та мітохондріальними. Хвороба вважається досить розповсюдженою, якщо її частота складає 1 на 10000 новонароджених. При частоті 1 уражений на 11000–40000 новонароджених патологія має середню розповсюдженість.
5.4.1. Аутосомно-домінантні патології
На цей час ідентифіковано близько 1900 аутосомно-домінантних захворювань Найбільш відомими аутосомно-домінантними захворюваннями є хорея Гентинґтона, синдром Марфана, синдром Холта – Орама, нейрофіброматоз, ахондроплазія та інші. Характерною ознакою цих патологій є порушення синтезу структурних або специфічних білків (наприклад, гемоглобіну). Дія мутантного гена проявляється практично завжди. Хворі хлопчики та дівчатка народжується з однаковою частотою.
Хорея Гентинґтона зустрічається приблизно з частотою від 1:10000 до 1:20000. Мутантний ген HD-huntingtin, який викликає це захворювання, локалізований в короткому плечі 4-ої хромосоми (4p16.3) (мал. 38). Мутація полягає у збільшенні кількості триплетних повторів (ЦАГ) ділянки гена, яка кодує кінцеву частину молекули білка хантинґтину, функція якого поки-що невідома. У нормі кількість повторів варіює в межах від 11 до 34 триплетів. У хворих це число може бути від 37 до 100 і більше. Чим більше повторів має мутантний ген, тим раніше починається хвороба.
Чоловіки хворіють частіше, ніж жінки. У основі патології лежить прогресуюче ураження мозкових клітин, переважно базальних ядер (смугасте тіло). При цьому мозок хворого за розмірами скорочується приблизно на 20– 30%.
Характерними ознаками хвороби є неритмічні хаотичні мимовільні скорочення м’язів різних частин тіла та розлад поведінки. Захворювання може починатися з будь-якого з цих симптомів або з обох відразу. Воно може розвинутися в будь-якому віці, але найчастіше перші симптоми його з’являються в 30-50 років.
Хорея Гентинґтона починається поволі. Першими симптомами можуть бути непосидючість, метушливість рухів, які проте не розцінюється хворим та його родичами як захворювання. Проте з часом рухові аномалії наростають і можуть привести до інвалідності. Характерні часті, раптові неритмічні судомні рухи кінцівок або тулуба. Можливі спазми м’язів обличчя, схлипування, порушення мови. Порушується координація рухів при ходьбі: хода стає танцювальною (хореїчною). Пам’ять не погіршується аж до пізніх стадій захворювання, проте увага, мислення та виконавські функції порушуються вже на самому початку захворювання. Часто спостерігається пригніченість, байдужість, відчуженість, дратівливість, втрата контролю над поведінкою. В деяких випадках розвивається маячня та нав’язливі стани, у зв’язку з чим помилково діагностується шизофренія.
Тривалість захворювання різна, але в середньому складає 15 років. При ранньому початку (до 20 років) патологія супроводжується стійким підвищенням м’язового тонусу, порушеннями координації рухів та мислення і прогресує швидше (середня тривалість складає 8 років). При цьому можливі часті епілептичні напади.
У більшості випадків хорея Гентинґтона виявляється в 40–50-річному віці прогресуючими мимовільними рухами, які супроводжуються судомами м’язів, а також вираженими психічними розладами (порушеннями пам’яті, пригніченістю, спробами самогубства, втратою емоційного контролю з частими спалахами роздратування та агресії).
Хорея Гентинґтона супроводжується двома складними проблемами.
По-перше, ознаки патології звичайно виявляються в середньому віці, коли численні хворі вже мають дітей. Після появи симптомів тривалість життя складає в середньому близько 15 років, і це повільне згасання є додатковим джерелом переживань для хворих та їх рідних.
По-друге, оскільки ген, що кодує хорею Гентинґтона, домінантний, він завжди буде проявлятися. Так, якщо уражений один із батьків, вірогідність народження хворою дитини складає 50%.
Специфічне лікування захворювання відсутнє. Розлади рухової активності та поведінки зменшують застосуванням деяких медичних препаратів.
Синдром Марфана полягає у системному ураженні сполучної тканини і характеризується високою пенетрантністю та різною експресивністю. Частота його складає 1:10000-20000. Хвороба спричинена мутацією гена FBN1, локалізованого у довгому плечі хромосоми 15 (15q21.1) (мал. 38). Виявлена велика кількість мутацій цього гена, що спричинює значну клінічну поліморфність хвороби. Ген FBN1 кодує синтез білка фібриліну, що входить до складу сполучної тканини і забезпечу її пружність. Блокування синтезу цього білка веде до підвищеної розтяжності сполучної тканини.
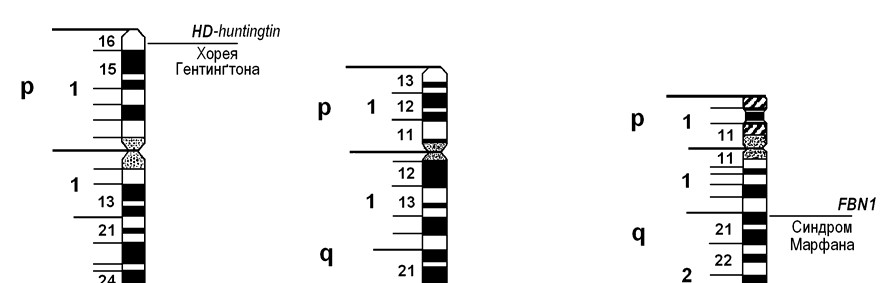
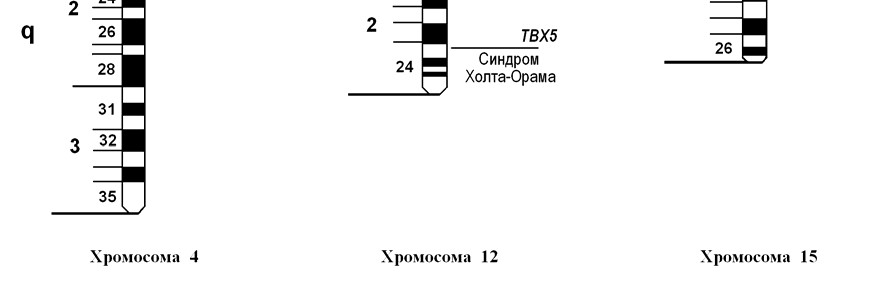
Мал. 38. Локалізація генів деяких аутосомно-домінантних патологій людини
Синдром Марфана уражує опорно-рухову, серцево-судинну системи та органи зору. Хворі мають характерний зовнішній вигляд: високий зріст, астенічну (кволу, слабку) статуру (мал. 39). Порушення опорно-рухової системи включають непропорційно довгі пальці (арахнодактилія – «павукові» пальці), видовжений череп, деформацію грудної клітки (воронкоподібна або кілеподібна), викривлення хребта, надмірна рухомість суглобів, плоскостопість. Найбільш характерними порушеннями серцево-судинної системи є випинання мітрального клапана в бік лівого передсердя, розширення аорти у висхідному або черевному відділі з розвитком аневризми (випинання). Патологія органів зору полягає у короткозорості високого ступеня внаслідок підвивиху (або зсуву) кришталика та різному кольорі райдужки. Можуть бути також пахові, стегнові, діафрагмальні грижі. Іноді трапляється опущення нирок, емфізема легенів (див. термінологічний словник), ослаблення слуху аж до повної глухоти. Попри всі ці порушення психічний та розумовий розвиток хворих відповідає нормі.
Тривалість життя хворого на синдром Марфана визначаються ступенем ураження серцево-судинної системи і сягає в середньому 35 років.
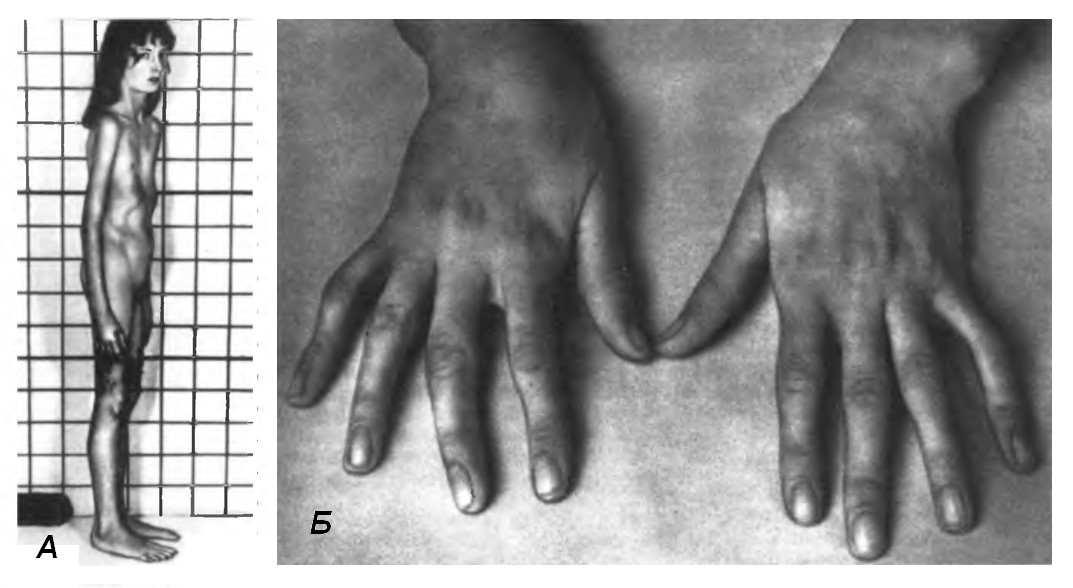
Мал. 39. Синдром Марфана:
а) непропорціональна статура, б) арахнодактилія
Лікування переважно симптоматичне: вживаються ліки для уповільнення руйнування аорти, а також гормональні препарати для стимуляції пропорційного статевого дозрівання дівчат. Позитивний вплив показує масаж, лікувальна гімнастика, інколи реконструктивна серцево-судинна хірургія.
Синдром Холта – Орама (синдром «рука – серце») супроводжується множинними природженими вадами розвитку. Частота захворювання покищо не визначена. Мутації гена ТВХ5, який розташований в довгому плечі хромосоми 12 (12q24.1), призводять до відсутності його продукту, внаслідок чого розвивається хвороба (мал. 38).
Клінічна картина синдрому Холта – Орама характеризується аномаліями верхніх кінцівок і природженими вадами серця. Вади розвитку рук варіюють від недорозвитку чи відсутності 1-го пальця кисті або його трьохфаланговості до недорозвитку або повної відсутності променевої кістки з формуванням променевої косорукості. Частіше вражається ліва рука. Спостерігаються і інші скелетні зміни: недорозвиток лопаток і ключиць, сколіоз (бокове викривлення хребта), воронкоподібна деформація грудини, викривлення мізинця, зрощення пальців, недорозвиток інших пальців кисті. У 50% хворих 1ий палець не протиставлений решті пальців кисті. (Мал. 40).
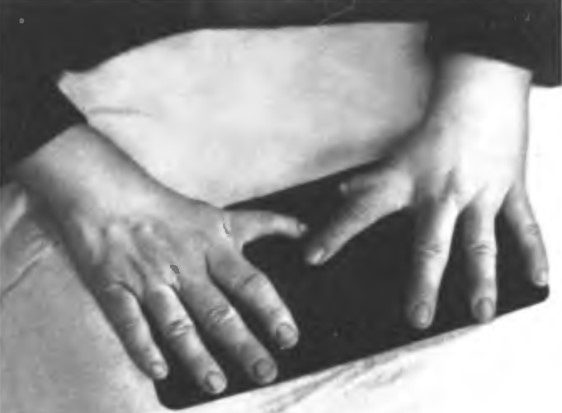
Мал. 40. Аномалії великих пальців рук у хворого на синдром Холта – Орама
У переважної більшості хворих (до 85%) виявляються різні форми природжених вад серця: дефекти міжпередсердної та міжшлуночкової перегородок, відкрита артеріальна протока (за нормою наявна в кровоносній системі плоду), звуження аорти та легеневої артерії, випинання мітрального клапана в бік лівого передсердя тощо.
Інтелект хворих на синдром Холта – Орама, як правило, зберігається.
Прогноз життя залежить від тяжкості ураження серця.
Лікування синдрому Холта – Орама полягає у медикаментозному запобіганні розвитку інфекційних хвороб серця (наприклад, ендокардиту) та реконструктивній хірургії серцевих перегородок чи клапанів.
5.4.2. Аутосомно-рецесивні патології
Відомо до 1900 аутосомно-рецесивних патологій, характерною ознакою яких є порушення функції одного чи кількох ферментів. Такі хвороби називаються ферментопатіями, або ензимопатіями. Найбільш поширеними серед них є .муковісцидоз, фенілкетонурія, галактоземія, хвороба Гоше, адреногенітальний синдром та інші. Дія мутантного гена проявляється лише у гомозиготному стані. Хворі хлопчики та дівчатка народжуються з однаковою частотою.
Муковісцидоз. Спадкове захворювання, обумовлене системним ураженням усіх екзокринних залоз організму – бронхолегеневої системи, кишечника, підшлункової залози, жовчної системи печінки, слинних, потових, слізних залоз, що призводить до утворення в’язкого секрету. В’язкі виділення закупорюють протоки залоз, накопичуються там і утворюють кісти (патологічні порожнини), що спричиняє порушення їх функцій.
Муковісцидоз – одне з найтяжчих та найпоширеніших моногенних захворювань дитячого віку. Частота захворювань для країн Європи та Північної Америки варіює в межах 1 на 2000-4000 новонароджених; у країнах Азії зустрічається рідко.
Ген муковісцидозу CFTR локалізований в 32-ому сегменті довгого плеча 7-ої хромосоми (7q32) і кодує білок-регулятор трансмембранної провідності іонів хлору (мал. 41). У даний час відомо понад 900 патологічних мутацій цього гена, переважно делецій трьох пар основ. У випадку гомозиготності мутантних алелей цього гена аніони хлору затримуються в епітеліальних клітинах, підсилюють поглинання катіонів натрію та води, спричинюючи «висушування» секретів екзокринних залоз.
Клінічні прояви хвороби розвиваються лише у гомозигот по аномальному гену. У гетерозиготних носіїв цього гена звичайно не виявляється ніяких симптомів захворювання.
Якщо обоє батьків є носіями дефектного алеля гена CFTR, то вірогідність народження дитини з муковісцидозом за кожної вагітності рівна 25%.
При цьому половина дітей може стати носіями аномального гена.
Якщо носієм гена CFTR є тільки один із батьків, то половина дітей вірогідно будуть гетерозиготними носіями цього гена, а небезпека народження хворої дитини відсутня.
Для дітей, уражених муковісцидозом, характерна схильність до повторних бронхітів, пневмоній, розвитку спадання частини легені, до хронічних кишкових захворювань, запалення підшлункової залози, запорів, випадання прямої кишки. При цьому спостерігається погане сприймання жирної їжі, рідкі та часті випорожнення, затримка фізичного розвитку. У чоловіків з часом може виявитися безплідність. Середня тривалість життя хворих на муковісцидоз складає близько 30 років.
Для діагностування муковісцидозу користуються аналізом поту на підвищений вміст іонів натрію та хлору та іншими клінічними показниками функціональних порушень дихальної та травної систем.
Лікування муковісцидозу переважно симптоматичне. При цьому застосовують засоби, що розріджують мокроту, ферментні препарати для поліпшення перетравлювання жирів їжі. Для боротьби з інфекцією застосовуються антибіотики з попереднім визначенням чутливості виділеного збудника. Використовуються також фізіотерапевтичні методи. Одним із видів фізіотерапії є лікувальна фізкультура, яка включає цикл активного дихання, аутогенний та руховий дренаж легень у поєднанні з перкусивним масажем (постукування пальцями рук). Метою лікувальної фізкультури є видалення мокроти з бронхіального дерева. Інколи при необхідності застосовують хірургічне втручання (кишкова непрохідність, накопичення повітря чи газів у порожнині плеври легень, пересаджування органів – легень, печінки, підшлункової залози).
Дієта хворого не повинна бути обмеженою. Калорійність харчування має досягати 120-150% від нормальної, причому 35% за рахунок жирів.
Обов’язковим є додаткове вживання вітамінів А, D, Е, К.
У даний час активно розробляються методи генної терапії муковісцидозу. Освоєна технологія клонування ДНК нормального гена. Доведено, що її введення в культуру уражених клітин усуває дефект мембранних каналів.
Найбільш вірогідною тканиною-мішенню є епітелій дихальних шляхів.
Розробляються системи перенесення генів на основі векторів – аденовірусів та ліпосом. До проблем, пов’язаних з генотерапією, відносяться дуже низький рівень перенесення генної конструкції в епітеліальні клітини, низький рівень експресії упровадженого гена та її скороминущий характер, розвиток імунної відповіді на білок вектора як антитілами, так і фагоцитами розвиток як місцевих так і системних запальних реакцій.
Одним з ефективних способів профілактики муковісцидозу є молекулярно-генетична пренатальна діагностика в сім’ях високого ризику.
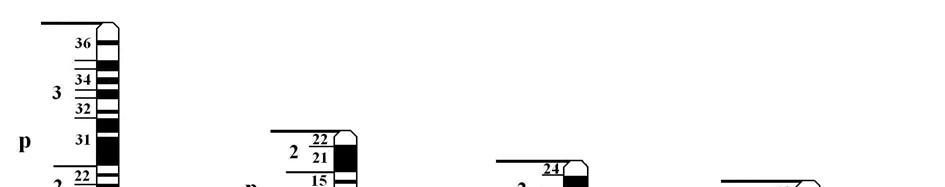
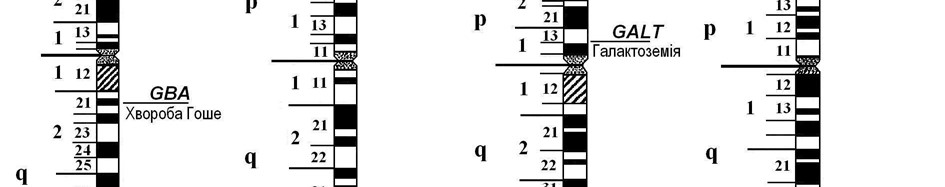
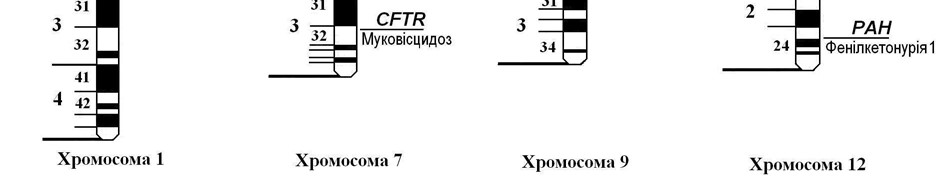
Мал. 41. Локалізація генів деяких аутосомно-рецесивних патологій
Фенілкетонурія. Серед новонароджених частота фенілкетонурії складає приблизно 1:10000, а серед розумово відсталих дітей – 1:1000.
Як відомо, білки їжі в шлунково-кишковому тракті розщеплюються до амінокислот, які всмоктуються в кров. За нормою амінокислота фенілаланін під впливом ферменту фенілаланін-4-гідроксилази, що утворюється в печінці, перетворюється на амінокислоту тирозин. Синтез цього ферменту здійснюється геном РАН, який розташований у 24 сегменті довгого плеча 12-ої хромосоми (12q24). Сьогодні відомо близько 200 мутацій цього гена, кожна з яких спричинює фенілкетонурію. (Мал. 41).
Може трапитися, що у подружній парі і чоловік, і жінка мають дефективний алель цього гена. Самі вони не страждають від нестачі цього ферменту, оскільки у кожного з них на гомологічній хромосомі знаходиться нормальний алель гена РАН. Однак, коли дитина від кожного із таких батьків успадковує аномальний ген, у неї розвивається фенілкетонурія. При цьому необхідний фермент або не виробляється зовсім, або має дуже слабку активність. У крові хворого нагромаджується велика кількість амінокислоти фенілаланіну та продуктів її напіврозпаду (фенілпіровиноградна, фенілоцтова кислоти тощо), які токсичні для організму, отруюють нервову систему дитини, шкідливо діють на інші органи і тканини. В даний час молекулярний механізм даної патології досліджено досить добре.
Хвороба супроводжується виразною затримкою психічного розвитку дитини, яка, як правило, абсолютно не засвоює найпростіші поняття, не може навчитися розмовляти і не розуміє мови. Перші ознаки хвороби з’являються в 2–6-місячному віці. Ранніми симптомами є запах цвілі («мишачий» запах), який має сеча та шкіра хворої дитини, напади блювання та загальне збудження. Характерними ознаками хвороби є також зниження м’язового тонусу, судомні напади. Як правило, діти, хворі на фенілкетонурію – блакитноокі блондини зі світлою шкірою та вираженими проявами алергії слизових оболонок.
З перших днів життя в крові такої дитини підвищений рівень фенілаланіну, а з сечею виділяється надмірна кількість фенілпіровиноградної та інших кислот. Саме ці показники використовують для діагностування фенілкетонурії
Лікування фенілкетонурії здійснюється шляхом призначення малобілкової дієти, що обмежує надходження фенілаланіну з їжею до мінімальної вікової потреби. В харчовий раціон хворих вводять овочі, фрукти, соки, а також спеціальні продукти з низьким вмістом білка. Особлива увага надається додатковому вживанню вітамінів, мінеральних речовин та мікроелементів.
Дієтотерапія призначається на тривалий термін (мінімум до 8-10 років).
Під контролем лікаря проводиться лікування, спрямоване на стимуляцію розвитку нервової системи дитини.
Якщо лікування почати не пізніше двомісячного віку, то в більшості випадків розвиток дитини йде практично нормально. В процесі дорослішання в організмі хворого формуються механізми, які протистоять патології, і він при проведенні певної корекції в харчуванні, може вести звичайний спосіб життя.
Галактоземія. Ще одним захворюванням, при якому дефект одного гена приводить до серйозних біохімічних змін в організмі, що викликають порушення розвитку і навіть загибель дитини, є галактоземія. Вона трапляється у 1 дитини на 15-20 тисяч новонароджених
Галактоземія спричинюється гомозиготною комбінацією аномальних алелей гена GALT, який локалізований в 13-му сегменті короткого плеча 9-ої хромосоми (9p13) (мал. 41). Відомо понад 50 аномальних мутацій цього гена, переважно у вигляді замін нуклеотидів.
Захворювання виявляється з перших місяців життя дитини і пов’язане з вигодовуванням грудним або коров’ячим молоком. Відомо, що основним вуглеводом у молоці є молочний цукор – лактоза. Лактоза в шлунковокишковому тракті розщеплюється на два моносахариди – галактозу та глюкозу. Проте клітинами організму використовується тільки глюкоза. Галактоза ж у нормі за допомогою ферменту галактозо-1-фосфат-урідилтрансферази також перетворюється на глюкозу. При галактоземії виявляється відсутність цього ферменту. В результаті в крові накопичується велика кількість галактози, яка отруйно діє практично на всі органи та тканини тіла дитини. Вигодовування дитини молоком досить швидко веде до розладу травлення, збільшення печінки, затримки розумового та психічного розвитку. Дитина жовтіє і різко худне, розвивається помутніння кришталика, що спричинює сліпоту. Гострі форми галактоземії призводять до смерті у перші місяці життя дитини.
Діагностується галактоземія через визначення активності ферменту галактозо-1-фосфат-урідилтрансферази в еритроцитах і концентрації галактози в крові та сечі.
Основний метод лікування хвороби полягає у призначенні низьколактозної дієти. Якщо хворій дитині з перших тижнів життя не давати молока, то вона розвивається нормально. Для харчування таких дітей розроблені спеціальні безлактозні суміші на основі соєвого або мигдалевого молока. При цьому дієта повинна включати достатню кількість овочів, фруктів, а також м’ясо, сало, крупи тощо.
Крім дієти використовуються ліки, які стимулюють функції нервової та кровоносної систем. При необхідності застосовується хірургічне втручання.
Хвороба Гоше (цереброзидоз). Хвороба Гоше не вважається розповсюдженою хворобою (частота її складає 1:40-60 тис. новонароджених), але займає серед спадкових ензимопатій особливе місце, бо є прикладом успішного розвитку досліджень таких патологій. Так, для хвороби Гоше визначено первинний біохімічний дефект, досліджені структури нормального білка і нормального гена, розроблені та впроваджені в практику методи ферментозамінної терапії, а в окремих випадках застосовуються трансплантації кровотворних клітин, визначені напрямки генної терапії.
Хвороба Гоше обумовлена мутацією гена GBA, локалізованого в 21-му сегменті довгого плеча 1-ої хромосоми (1q21) (мал. 41). Ген GBA контролює синтез ферменту бета-D-глюкоцереброзидази, який бере участь у розщепленні глюкозилцераміду на глюкозу та церамід. Дослідження патології на генному рівні ускладнюється значною кількістю (понад 100) різноманітних мутацій гена, які відрізняються різною активністю ферменту глюкоцереброзидази.
Наслідком мутації гена є недостатня активність глюкоцереброзидази, в результаті чого глюкозилцерамід накопичується в лізосомах лейкоцитів, здатних до фагоцитозу. Розміри таких клітин непомірно збільшуються (клітини Гоше). Клітини Гоше утворюються в тканинах головного мозку, печінки, селезінки, червоного кісткового мозку, лімфатичних вузлів та інших органів, що є характерною ознакою даної патології. Нагромадження цереброзиду в клітинах нервової системи спричиняє її руйнування.
Розрізняють дитячу та юнацьку форми хвороби Гоше. Дитяча форма проявляється в перші місяці життя затримкою фізичного та розумового розвитку, збільшенням живота, печінки та селезінки, ускладненням ковтання спазмами горлянки. Можлива дихальна недостатність через ущільнення легеневої тканини, судоми. Смерть хворої дитини наступає протягом першого року життя. (Мал. 42).
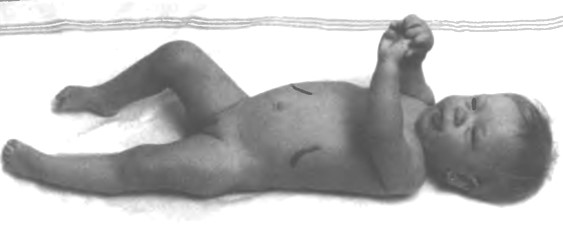
Мал. 42. Хвороба Гоше
Діагностується хвороба Гоше на підставі визначення активності ферменту бета-D-глюкоцереброзидази в лейкоцитах хворого та одного з батьків, а також у фібробластах (клітини, що утворюють волокна) шкіри хворого.
Крім того, здійснюють аналіз ДНК лейкоцитів хворого та одного із батьків.
До недавнього часу медицина не володіла ефективними засобами лікування уражених хворобою Гоше. Терапія носила переважно частковий характер (видалення селезінки, трансплантація кісткового мозку, обмеження рухової активності тощо).
Останнім часом дане захворювання стало предметом міждисциплінарного вивчення з боку генетики, ортопедії, гематології та молекулярної біології, що привело до обґрунтовування та використання нових методів лікування. В даний час лікування хвороби Гоше, на відміну від інших генетичних захворювань, вважається високоефективним. Гарні результати показує ферментозамінна терапія – регулярне введення в кров ферменту бета-Dглюкоцереброзидази, який ефективно виконує свою функцію.
5.4.3. Патології, зчеплені зі статтю
Відомо понад 650 хвороб, зчеплених (або гадано зчеплених) з Ххромосомою. Тяжкість захворювання залежить від статі. Повний прояв хвороби спостерігається переважно у чоловіків, оскільки вони гемізиготні (напівзиготні) за генами, локалізованими в Х-хромосомі. Якщо патологічний ген
є рецесивним, то гетерозиготні жінки здорові, але є носіями цього гена, а гомозиготні жіночі ембріони переважно летальні. У випадку домінантності аномального гена гетерозиготні жінки уражені легкою формою хвороби, а гомозиготи летальні. У чоловіків тільки одна Х-хромосома, тому у них Хзчеплена хвороба частіше виявляється повністю, незалежно від того, домінантна вона у жінок чи рецесивна.
Таким чином, поняття домінантного та рецесивного Х-зчеплення стосується звичайно прояву ознаки у жінок. Хоча і тут часто відсутні чіткі визначення домінантності чи рецесивності ряду хвороб.
Важливою особливістю Х-зчепленого успадкування є те, що ознака не передається по чоловічій лінії, оскільки син одержує від батька Y-хромосому. Але всі дочки батька, ураженого Х-зчепленою хворобою, успадковуватимуть аномальний алель гена, оскільки вони обов’язково одержують від батька Ххромосому.
Х-зчеплені домінантні захворювання. Ці патології зустрічаються рідше, ніж рецесивні. Найбільш відомими серед них є гіпофосфатемічний рахіт, синдром Блоха – Сульцбергера (пігментне нетримання), осередкова мезоектодермальна дисплазія, окремі форми нефрогенного нецукрового діабету, темна емаль зубів та деякі інші. Хворіють як чоловіки, так і жінки.
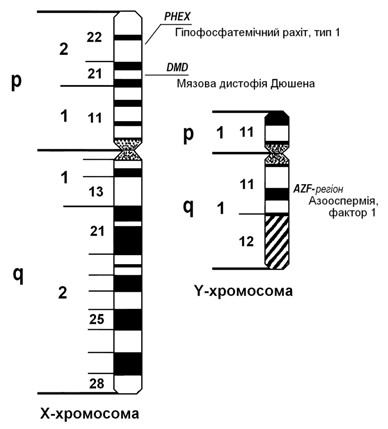
Мал. 43. Локалізація генів деяких патологій, зчеплених зі статтю Гіпофосфатемічний рахіт тип 1 – найпоширеніше захворювання серед дитячих рахітів, частота якого складає 1:25000. Воно зумовлене мутацією гена PHEX, локалізованого на короткому плечі Х-хромосоми (Хр22.1), який контролює реабсорбцію (зворотне поглинання) фосфатів у ниркових канальцях та в кишечнику (мал. 43). Патологія полягає у різкому зменшенні концентрації неорганічного фосфору в крові (гіпофосфатемія), що може призвести до порушення процесу перетворення неактивної форми вітаміну D в активну. Хлопчики хворіють тяжче, ніж дівчатка, бо вони гемізиготні, а дівчатка гетерозиготні за цим геном. У матерів деяких хворих спостерігаються клінічні ознаки патології у вигляді деформації кісток або низького зросту, у інших виявляється лише гіпофосфатемія натщесерце.
Перші ознаки хвороби виразно стають помітні на початку 2-го року життя або пізніше, коли дитина починає ходити. Звертають на себе увагу хитка («качина») хода, зменшення рухової активності, низькорослість, наростаюче ) (– або ( )-подібне викривлення гомілок і менш виражена деформація решти частин скелета (мал. 44). Іноді дитина перестає ходити через болі в кістках. У хворих можливі спонтанні переломи.
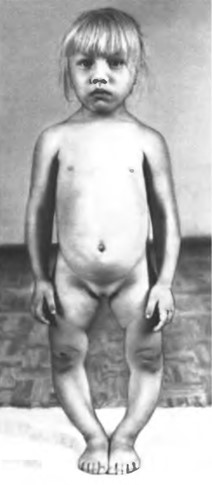
Мал. 44. Гіпофосфатемічний рахіт
Психічно дитина розвивається нормально, але стає мовчазною, не спілкується з однолітками. У дівчаток хвороба проявляється у легкій формі – лише затримкою росту та гіпофосфатемією. Іноді захворювання у дитини проходить без лікування, але поновлюється в дорослому віці, наприклад у жінок під час вагітності та лактації.
Лікування загальновизнаними дозами вітаміну D виявляється безуспішним. Проте хвороба відступає під дією великих доз активних метаболітів цього вітаміну. В лікувальний комплекс включають також препарати кальцію та фосфору. У випадку тяжких уражень скелета застосовують ортопедичні засоби, а якщо необхідно, то і хірургічне втручання. З метою запобігання деформації кісток немовляті не дають надто рано ставати на ноги та ходити.
Х-зчеплені рецесивні захворювання. На ці хвороби нездужають переважно чоловіки. Тяжкість таких патологій варіює в широких межах, наприклад у чоловіків – від безпліддя за синдрому Леша – Найхана до порівняно легких порушень у вигляді деяких форм облисіння. До Х-зчеплених рецесивних захворювань відносяться міодистрофія Дюшена, гемофілія (див. розділи 3.3.3. та 4.3.2.), ангідротична ектодермальна дисплазія, вроджена гіпоплазія наднирникових залоз, адренолейкодистрофія, дальтонізм та інші.
М’язова дистрофія Дюшена є досить розповсюдженою та тяжкою нервово-м’язовою хворобою. Частота її складає 1:3000-5000 новонароджених хлопчиків. Захворювання обумовлене порушенням синтезу білка дистрофіну, ген якого (DMD) локалізований в короткому плечі Х-хромосоми (Xp21.2) (мал. 43).
Перші ознаки хвороби, у вигляді утруднень при вставанні з підлоги чи після присідання, з’являються у 3-5 років. Хворі діти не вміють бігати та скакати. Захворювання полягає у прогресуючій дистрофії м’язів з поступовим до 14-15 років знерухомленням дитини. М’язова тканина заміняється жировою та сполучною, починаючи з м’язів ніг. Ознаки хвороби розповсюджуються поступово угору по тілу. При цьому порушується моторика шлунковокишкового тракту. На завершальній стадії хвороби атрофія захоплює м’язи обличчя, глотки та дихальні м’язи, порушуються функції серця. Хворі гинуть протягом 2-3-го десятиліття життя.
У половини хворих можливе зниження інтелекту аж до дебільності.
Лікування, спрямоване на підтримання фізичної активності (спеціальна лікувальна гімнастика) пацієнта, та покращення умов життя виявляються малоефективними. Використовують протези, які дозволяють хворим рухатися та уповільнюють формування сколіозу. При появі обмеження рухливості суглобів показане хірургічне втручання. Для уповільнення розвитку хвороби призначають медикаментозне лікування.
Y-зчеплені хвороби. Цих патологій відомо всього кілька, оскільки чоловіча статева хромосома має незначну кількість генів (близько 100), із яких ідентифіковано близько 20. Гени, які визначають такі патології, не мають аналогів у Х-хромосомі. Таким чином, Y-зчеплені хвороби визначаються лише одним алелем, виявляються тільки у чоловіків і передаються від батька усім синами. Серед цих захворювань – азооспермія, гіпертрихоз (волосатість країв ушних раковин), перетинки між пальцями ніг та іхтіоз (лускоподібна шкіра).
Азооспермія. На Y хромосомі людини розташовано кілька десятків генів, контролюючих формування статевих ознак, у тому числі розвиток яєчок та процес сперматогенезу. Різні мутації в структурі цих генів, як правило, приводять до порушення розвитку статевої системи. Основні гени, контролюючі сперматогенез у чоловіків, розташовані в певній ділянці довгого плеча Y-хромосоми (Yq11) (мал. 43). Цю ділянку прийнято називати AZF-регіон (від англ. azoospermic factor). Його підрозділяють на три субрегіони – AZFa, AZFb та AZFc. У кожному з цих субрегіонів знаходяться різні гени, мутації яких ведуть до широкого спектру порушень сперматогенезу – від недостатньої кількості сперматозоонів у спермі (олігоспермія) до їх повної відсутності (азооспермія).
Частота розповсюдження спадкової азооспермії варіює в межах 0,170,24%. Вона виникає в результаті мікроделецій у локусі AZF Y-хромосоми.
Найтяжчими є мікроделеції в субрегіонах AZFa та AZFb.
На даний момент ефективних засобів лікування спадкової азооспермії не існує, і вірогідність відновлення фертильності для хворих дуже мала. Однак, це не значить, що їх сім’яники не здатні виробляти хоча б незначну кількість нормальних гамет. Проблема в іншому: поки-що не існує технології, яка б забезпечила сепарацію зрілих сперматозоонів із їх загальної маси з наступним штучним заплідненням. Крім того, досить часто від сперми чоловіків з порушеннями локуса AZF формуються нежиттєздатні ембріони.
Статеві хромосоми людини X та Y мають також гомологічні ділянки з алельними генами, які з однаковою вірогідністю наявні у обох статей. Ознаки, детерміновані цими генами, успадковуються за правилами Г. Менделя, як звичайні аутосомні ознаки.
До рецесивних патологій, які визначаються алельними генами X- та Y- хромосом, належать одна із форм пігментної ксеродерми, гонадний дисгенез та загальна кольорова сліпота.
Пігментна ксеродерма. Внаслідок спадкового дефекту ферментів хеліказ, які беруть участь у репарації (відновлення) пошкоджень ДНК, має місце послаблення або повна відсутність репаративних процесів у ДНК при ушкоджувальному впливові різноманітних мутагенних факторів, внаслідок чого у людини під дією УФ-променів сонячного світла розвивається досить рідкісна хвороба – пігментна ксеродерма. Популяційна частота її складає 1:250000. Відомі також інші назви цієї патології: пігментна атрофія шкіри Брокера, атрофія шкіри з меланозом і телеангіектазіями Нейсера, прогресуючий лентикулярний меланоз Піка.
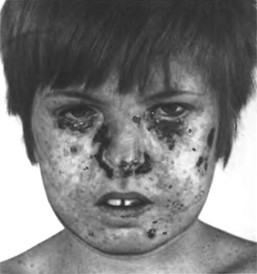
Мал. 45. Пігментна ксеродерма
При пігментній ксеродермі на відкритих частинах тіла у дітей віком від 3 місяців до 3 років з’являються пігментні плями, які поступово перетворюються на пухлини (мал. 45). Хвороба має несприятливий прогноз, бо звичайно спостерігається летальний наслідок.
Ген цієї форми пігментної ксеродерми та його локалізація поки-що не визначені.
Хворі повинні уникати впливу на шкіру сонячного проміння, використовувати сонцезахисні креми. Для лікування пігментної ксеродерми використовують спеціальні ароматичні та клітиновідновлюючі ліки.
5.4.4. Мітохондріальні хвороби
Мітохондріям належить провідна роль в утворенні енергії в результаті окислення вуглеводів, жирів та білків. Дефект будь-якого з ферментів мітохондрій порушує їх функцію. При цьому в першу чергу страждають найбільш енергозалежні тканини і органи – центральна нервова система, скелетні та серцевий м’язи, нирки, печінка, ендокринні залози. На фоні хронічного дефіциту енергії в них рано чи пізно виникають патологічні зміни і розвиваються захворювання, які одержали назву мітохондріальних. Сучасній медицині відомо близько 200 таких хвороб. В їх клініці зустрічається сама різні патології, але домінують ураження центральної нервової системи та м’язової тканини.
Симптомами, типовими для мітохондріальних захворювань, є м’язові болі, слабкість і атрофія мускулатури, нездатність до фізичних навантажень, опущення верхніх повік, ушкодження периферійних нервових волокон, судоми, відсутність рефлексів, атрофія зорового нерва, нейросенсорна глухуватість, мігрень, летаргійні стани, порушення психомоторного розвитку (рухові акти, трудова діяльність, навички та вміння), недоумкуватість, розлад пам’яті, уваги, мислення, мови та поведінки.
Мутації, що виникли в мітохондріальних генах, передаються в нові мітохондрії при поділі цих органел. Виходить, що навіть у межах однієї клітини наявні мітохондрії з різними варіантами геномів. Таким чином, людина з мутацією в мітохондріальному гені несе суміш нормальної та мутантної ДНК. При цьому співвідношення мітохондрій з мутантними та нормальними геномами може бути будь-яким, тому прояв мітохондріальних захворювань у різних хворих неоднаковий. У подібних випадках мутації спочатку можуть взагалі не мати зовнішніх ознак. Нормальні мітохондрії до певного часу забезпечують клітини енергією, компенсуючи недостатність функції мітохондрій з дефектами. Це проявляється різним за тривалістю безсимптомным періодом при багатьох мітохондріальних захворюваннях. Проте рано чи пізно наступає момент, коли дефектні мітохондрії нагромаджуються в кількості, достатній для прояву патологічних ознак. Вік маніфестації захворювання варіює у різних хворих. Ранній початок захворювання приводить до тяжчого його перебігу та несприятливого прогнозу.
Успадкування мутацій у мітохондріальному геномі носить особливий характер. Якщо гени, локалізовані в ядерній ДНК, діти одержують порівну від обох батьків, то мітохондріальні гени передаються нащадкам тільки від матері. Це пов’язано з тим, що всю цитоплазму з мітохондріями, що містяться в ній, нащадки одержують разом з яйцеклітиною, тоді як в сперматозоонах цитоплазма практично відсутня. З цієї причини жінка з мітохондріальним захворюванням передає його всім своїм дітям, а хворий чоловік – ні.
Кожний із відомих сьогодні синдромів, викликаних порушенням функціонування мітохондрій, визначається якою-небудь мутацією наступного типу: нуклеотидні заміни в генах, втрати або вставки фрагментів мтДНК, зміна числа копій мтДНК.
Сьогодні ідентифіковано понад 20 спадкових мітохондріальних патологій, до яких належать спадкова атрофія зорових нервів Лебера, пігментний ретиніт (точніша назва хвороби: нейропатія, атаксія та пігментний ретиніт), міоклональна епілепсія з надзвичайно червоними м’язовими волокнами, нейросенсорна глухота тощо.
Атрофія зорових нервів Лебера. Відомо принаймні 10 точкових мутацій генів, які пов’язані з синдромом Лебера. Вони спричинюють заміну тієї чи іншої амінокислоти у одного з ферментів-дегідрогеназ, що призводить до порушення його активності. При даному захворюванні у людей 20-30 років відбувається майже повна втрата центрального зору через атрофію зорових нервів та дегенерацію гангліозного шару клітин сітківки. Хворіють переважно чоловіки (80-85%). Виявлено, що у 95% випадків причиною патології є мутації в трьох мітохондріальних генах – ND1 (мутація LHON 3460 A), ND4 (мутація LHON 11778 A) та ND6 (мутація LHON 14484 C) (мал. 46).
Решта 7 мутацій, пов’язаних з хворобою Лебера, вважаються «вторинними» і можуть підсилювати дію первинних мутацій, збільшуючи ризик прояву захворювання.
Синдром Лебера – найпоширеніше з усіх відомих на даний час мітохондріальних спадкових захворювань – його частота в Європі складає близько 1:25000.
Лікування здійснюється препаратами, які поліпшують обмін речовин, розширюють судини, та вітамінами, а також фізіотерапевтичними засобами (електростимуляція, магнітотерапія тощо).
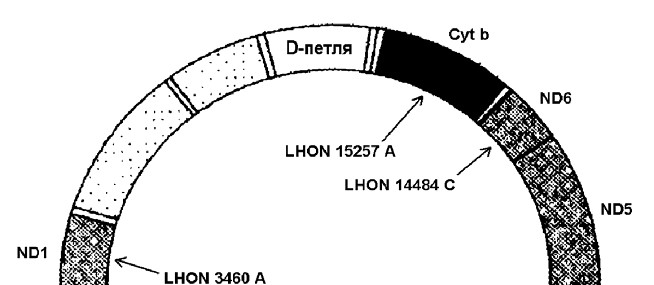
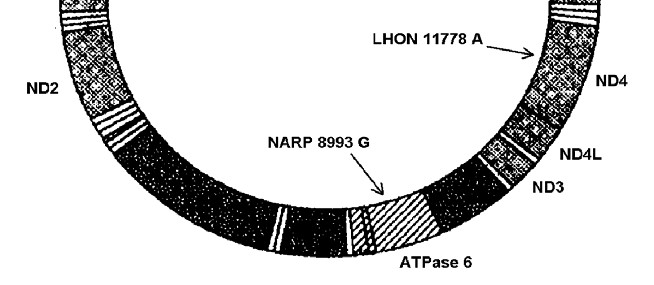
Мал. 46. Локалізація генів деяких мітохондріальних патологій (М-хромосома)
Нейропатія, атаксія та пігментний ретиніт. Цю комплексну хворобу викликає точкова мутація NARP 8993 G в гені АТРase 6, який кодує один із ферментів АТФ-синтетазного комплексу. (мал. 46). Захворювання розвивається за наявності в клітинах 70-90% аномальної мтДНК. Ознаками патології є затримка розвитку, розумова відсталість, прогресуюче звуження зорових полів та нічна сліпота, біль та порушення чутливості у відповідних зонах іннервації, розлад координації довільних рухів і нейрогенна м’язова слабкість. До речі, сьогодні відомо принаймні 15 форм пігментного ретиніту (прогресуюче звуження зорових полів та нічна сліпота), спричинених домінантними та рецесивними мутаціями генів, які локалізовані в ряді аутосом та Ххромосомі.
Якщо мутантна ДНК складає понад 90% від усієї мтДНК, то розвивається зовсім інша патологія – хвороба Лея, яка характеризується ураженням головного мозку, атрофією зорових нервів, зниженням тонусу м’язів, розладом координації довільних рухів, мимовільними ритмічними рухами очних яблук та обмеженням їх довільної рухомості.