Генетика людини: методичка
5.5. Хромосомні хвороби
До хромосомних захворювань відносяться такі патології, які спричинюються структурними порушеннями (абераціями) окремих хромосом або зміною їх кількості. На цей час їх відомо понад 700. Хромосомні хвороби виникають в результаті порушень процесу утворення гамет одного із батьків (нерозходження тієї чи іншої пари гомологічних хромосом у процесі мейозу). Такі аномалії майже не успадковуються (всього 3-5%), а виникають щоразу заново.
Характерно, що хромосомні порушення зумовлюють близько половини всіх викиднів та 7% мертвонароджених.
Загальною особливістю всіх форм хромосомних хвороб є множинність ураження. Це черепно-лицьові дефекти, вроджені вади розвитку внутрішніх органів і частин тіла, сповільнений внутрішньоутробний та постнатальний ріст і розвиток, відставання психічного розвитку, порушення функцій нервової, ендокринної та імунної систем. При кожній формі хромосомних хвороб спостерігається 30-80 різних відхилень від норми, які немов би перекривають інші форми. Ряд хромосомних хвороб характеризується лише певним поєднанням відхилень у розвитку, а не специфічними вадами, що і використовується в клінічній і патологоанатомічній діагностиці.
Патогенез хромосомних хвороб починається на ранній стадії ембріогенезу, тому при народженні всі основні вади розвитку вже в наявності (окрім вад розвитку статевих органів).
Виявилося, що найбільш специфічні для того або іншого синдрому ознаки обумовлені порушеннями генетичного матеріалу порівняно невеликих сегментів хромосом. Так, специфічні клінічні симптоми синдрому Дауна мають місце при трисомії окремого сегмента довгого плеча 21-ої хромосоми (21q22.1). Для розвитку синдрому Лежена при делеціях короткого плеча 5-ої хромосоми найбільш важлива середня частина сегменту (5р15). Характерні риси синдрому Едвардса пов’язані з трисомією сегменту хромосоми 18q11.
Доречно зауважити, що 2/3 хромосомних аномалій складають аномалії статевих хромосом. Частота синдромів, пов’язаних з порушеннями будови чи кількості хромосом Х та Y, складає 5:1000 новонароджених. Хромосомні ушкодження виявлені у 5-15% чоловіків з безпліддям або порушеннями сперматогенезу. При цьому 75% таких чоловіків мають аномалії статевих хромосом і 25% – аномалії аутосом.
Характерно, що у випадку аномалій статевих хромосом спостерігається відсутність численних вад розвитку та розумової відсталості, які типові для аномалій аутосом. За патології хромосом Х та Y лише 1% хворих мають затримку розумового розвитку. До того ж вона виражена менше, ніж при аномаліях аутосом.
Клінічний поліморфізм кожної хромосомної хвороби в загальній формі обумовлений генотипом організму і умовами середовища. Варіації в проявах патології можуть бути дуже широкими: від летального ефекту до незначних відхилень у розвитку. Так, 60-70% випадків трисомії 21-ої хромосоми закінчуються загибеллю на ембріональній стадії, в 30% випадків народжуються діти з синдромом Дауна з широко варіюючими клінічними проявами. За моносомії по Х-хромосомі (синдром Шерешевського-Тернера) розвивається лише 10% зародків (інші гинуть), а якщо враховувати ще доімплатаційну загибель зигот Х0, то живі новонароджені з синдромом ШерешевськогоТернера складають тільки 1%.
5.5.1. Хромосомні аберації
Патологій, спричинених порушеннями структури хромосом (переважно делеціями та дуплікаціями), на сьогодні клінічно і цитогенетично ідентифіковано близько 70. Серед них найбільш відомими є синдром Вільямса – Бейрена, синдром Лежена, синдром Вольфа – Хіршхорна, міопатія Шарко – Марі – Тус та деякі інші.
Синдром Лежена (синдром «котячого крику») зустрічається з частотою 1:40000-50000 живих новонароджених внаслідок делеції в короткому плечі 5-ої хромосоми (5р). Розмір втраченого фрагмента в різних випадках різний. Для розвитку клінічної картини хвороби має значення не розмір утраченого фрагменту, а конкретна незначна ділянка хромосоми. Більшість випадків делецій виникає заново.
Виразним симптомом цієї хвороби є характерний плач дитини, який нагадує нявчання кішки. Такий плач пов’язаний з аномальним розвиток гортані (її звуженням, м’якістю хрящів, набряклістю чи надзвичайною складчастістю слизової оболонки, зменшенням надгортанника) або голосових зв’язок. Захворювання супроводжується різкою затримкою фізичного та розумового розвитку, мікроцефалією (надто зменшена голова). Хворі мають своєрідний зовнішній вигляд: місяцеподібне лице з низько розташованими та деформованими вухами, непропорційно мала верхня щелепа, збільшена відстань між очима, монголоїдний розріз очей та епікант (вертикальна складка шкіри у внутрішньому куті очної щілини), косоокість (мал. 47). Спостерігаються аномалії опорно-рухової системи, гіпотонія (зниження тонусу) м’язів, клишоногість, плоскостопість, зрощення суміжних пальців ніг, численні вади серця. Більшість дітей вмирає досить рано і лише близько 10% досягає 10-річного віку.
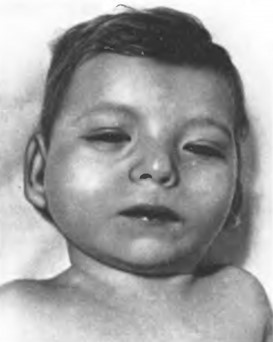
Мал. 47. Синдром Лежена
З віком такі діагностичні ознаки як «котячий крик», місяцеподібне обличчя та гіпотонія м’язів зникають зовсім, але мікроцефалія та розумова відсталість стають більш помітними.
Лікування симптоматичне: засоби, які стимулюють психомоторний розвиток, лікувальний масаж та гімнастика.
Синдром Вольфа – Хіршхорна спричинюється переважно делецією в короткому плечі 4-ої хромосоми (4р). Розмір делеції варіює від незначного до такого, що захоплює половину дистальної (крайньої) частини короткого плеча хромосоми. Виявлено, що переважна більшість делецій виникає заново. Поряд з делеціями патологію у новонароджених можуть викликати інверсії та дуплікації. Частота захворювання невелика – 1:100000 живих новонароджених.
Ознаками синдрому Вольфа – Хіршхорна є численні вроджені вади розвитку, затримка розумового та психічного розвитку. Вага хворих новонароджених невелика, не дивлячись на нормальну тривалість вагітності. Серед зовнішніх ознак характерними є мікроцефалія, дзьобоподібний ніс, епікант, опущені зовнішні кути очних щілин, деформовані вушні раковини, розщеплена верхня губа та піднебіння, маленький рот, деформовані стопи тощо.
Хворі діти мають низьку життєздатність і вмирають переважно до 1-го року.
5.5.2. Порушення кількості аутосом
Відомо близько 300 хвороб, спричинених порушеннями кількості аутосом. Найбільш відомими серед них є синдром Дауна, синдром Патау, синдром Едвардса, трисомія по 8-ій хромосомі та інші.
Синдром Дауна є найбільш вивченою хромосомною хворобою. Частота цього синдрому серед новонароджених складає 1:700-800 (0,12-0,14%) і не варіює за часовою, етнічною або географічною ознакою у батьків однакового віку. Звичайно загальна популяційна частота патології значно вища, бо понад 2\3 уражених плодів гине до народження. Частота народження дітей з синдромом Дауна залежить від віку матері (близько 80% випадків) і в меншій мірі від віку батька. Так, у віці 20 років вона складає в середньому 0,06%, а у віці 45 років – 3%. У надто молодих матерів (до 18 років) частота народження дітей з синдромом Дауна теж досить висока і складає близько 2%. Співвідношення хлопчиків і дівчаток серед хворих складає 1:1.
В переважній більшості випадків (до 94 %) у хворих виявляється проста трисомія 21-ої хромосоми (каріотип 47, XX (XY) + 21). Близько 4 % синдромів Дауна спричинені транслокаціями довгого плеча 21-ої хромосоми переважно на 14-у хромосому. Невелика (близько 2%) частка дітей з синдромом Дауна має мозаїчні форми (47+21/46).
Найголовніші діагностичні ознаки синдрому Дауна: монголоїдні очі (косо розміщені, з епікантом), широке перенісся, деформовані вушні раковини, напіввідкритий рот, збільшений язик, короткі шия, кисті та стопи тощо (мал. 48). Наявне також значне відставання у рості та статевому розвитку (хворі звичайно безплідні). Хвороба часто супроводжується вадами серця, органів травлення, неврологічними розладами (косоокість, мимовільні ритмічні коливальні рухи очних яблук, низький тонус м’язів тощо).





Мал. 48. Діти різного віку з характерними рисами синдрому Дауна
Психічні розлади виявляються у вигляді олігофренії на рівні дебільності (IQ = 51-70) або імбецильності (IQ = 21-50).
Медична допомога дітям з синдромом Дауна неспецифічна та різноманітна. Вроджені вади серця виправляють хірургічно. Хворі потребують постійного уважного догляду, загальнозміцнювального лікування, повноцінного харчування. Профілактика полягає у запобіганні застудних та інфекційних захворювань.
За відсутності виражених вад розвитку прогноз для життя відносно сприятливий.
Численні хворі на синдром Дауна в наш час здатні вести самостійне життя, оволодівають нескладними професіями, створюють сім’ї.
Синдром Патау по частоті розповсюдження (1:5000-7000 новонароджених) займає друге місце після синдрому Дауна серед аутосомних трисомій і також не залежить від статі. У переважній більшості випадків (80-85%) він спричинюється трисомією 13-ої хромосоми при каріотипі 47, ХХ (ХY) + 13. Решта випадків зумовлена транслокаціями її фрагментів на інші аутосоми.
Зовнішні ознаки синдрому настільки характерні, що дозволяють практично відразу після народження виявити це захворювання. Перш за все звертають на себе увагу мікроцефалія, скошений лоб, вузькі очні щілини, запале перенісся, низько розташовані та деформовані вушні раковини тощо. Найхарактернішими зовнішніми вадами розвитку є розщеплення верхньої губи та піднебіння, а також полідактилія. (Мал. 49).
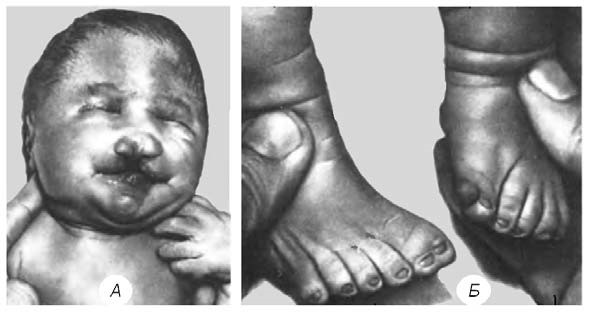
Мал. 49. Аномалії обличчя (А) та стопи (Б) за синдрому Патау:
У більшості хворих виявляються вроджені вади серця, органів травлення, нирок, статевих органів, органів зору. Ураження центральної нервової системи наявне в 100 % випадків, внаслідок чого всі діти з синдромом Патау відзначаються глибокою ідіотією (IQ = 0-21).
Лікування дітей, хворих на синдром Патау, неспецифічне: хірургічне втручання з нагоди вроджених вад розвитку, загальнозміцнювальні заходи, ретельний догляд, профілактика застудних та інфекційних захворювань.
Тривалість життя хворих дітей незначна – на першому році вмирає 95%. У віці понад 3 роки зустрічаються одиниці.
5.5.3. Порушення кількості статевих хромосом
Дотепер ідентифіковано понад 300 захворювань, спричинених порушеннями кількості статевих хромосом. Серед них найбільш відомими є синдром Клайнфельтера, синдром дисомії Y-хромосоми, синдром Шерешевського – Тернера, синдром трисомії Х-хромосоми та інші.
Синдром Клайнфельтера спричинюється каріотипом 47, ХХY. Зайву Х-хромосому хворий з рівною вірогідністю може отримати як від батька, так і від матері. Частота даної патології варіює в межах 0,5-2,0 на 1000 новонароджених хлопчиків. Поряд з цим зустрічаються варіанти з більшим числом Х- чи Y-хромосом, які також відносяться до синдрому Клайнфельтера.
Основні клінічні ознаки з’являються в період настання статевої зрілості. Класичними проявами цієї хвороби є високий зріст, євнухоподібна статура з вузькими плечима та широким тазом, гінекомастія (збільшення молочних залоз), але всі ці симптоми одночасно зустрічаються лише в половині випадків. Крім того у хворих спостерігається слабкий ріст волосся на обличчі, у пахвових западинах, на лобку, гіпогонадизм (недорозвиненість яєчок), який призводить до азооспермії та статевого інфантилізму (дитяча поведінка) (Мал. 50).
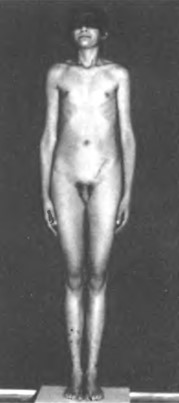
Мал. 50. Синдром Клайнфельтера
Супутні ознаки: деформація вушних раковин, алопеція (плішивість), катаракта, надмірний розвиток верхньої щелепи, вади серця тощо. Однак ці ознаки не мають діагностичного значення.
Близько 15% хворих на синдром Клайнфельтера мають ознаки легкої дебільності. У випадку значнішого порушення інтелекту спостерігається аутизм, недовірливість, схильність до алкоголізму, асоціальна поведінка.
Лікування симптоматичне гормональними препаратами.
Синдром Шерешевського – Тернера розповсюджений з частотою 1:2000-5000 новонароджених дівчаток.
Найбільш часто при цитогенетичному дослідженні виявляється каріотип 45, Х0, проте трапляються інші форми аномалій Х-хромосоми – делеції короткого або довгого плеча, ізохромосома (моноцентрична хромосома з обома гомологічними плечима), а також різні варіанти мозаїчності.
Дитина з синдромом Шерешевського – Тернера народжується тільки у разі неодержання Х-хромосоми від батька. При неодержанні Х-хромосоми від матері ембріон гине на ранніх стадіях розвитку.
Основними діагностичними ознаками цієї хвороби є низький зріст, набряк кистей та стоп, двостороння шкірна складка на шиї, низьке розташування та деформація вушних раковин, широка грудна клітка з широко розміщеними сосками, вроджені вади серця та нирок, недорозвиненість молочних залоз та яєчників, відсутність менструацій, статевий інфантилізм (мал. 51). Інколи спостерігається розумова відсталість та психопатія (неадекватна поведінка та недостатня соціальна адаптація). Тривалість життя хворих на синдром Шерешевського-Тернера наближається до норми.
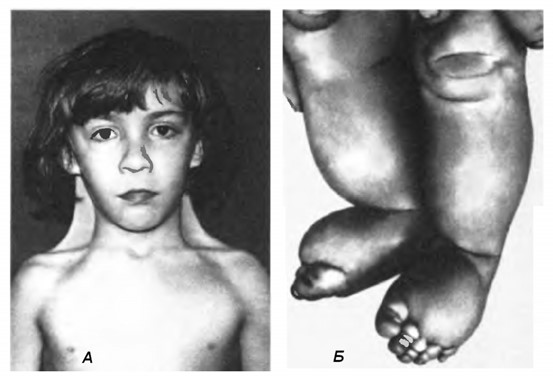
Мал. 51. Синдром Шерешевського – Тернера:
А – крилоподібні складки шкіри на шиї,
Б – характерні лімфатичні набряки на ногах
При мозаїчних формах клінічні прояви хвороби мінімальні.
Лікування комплексне: реконструктивна (вроджені вади внутрішніх органів) та пластична (крилоподібні складки тощо) хірургія, гормональні препарати, психотерапія.