Генетика людини: методичка
5.7. Спадкова схильність до хвороб
Поряд із хворобами, чітко детермінованими спадковістю (генами та хромосомами) або чинниками середовища (травми, опіки), є велика і різноманітна група хвороб, розвиток яких визначається взаємодією певних спадкових факторів (мутацій або поєднань алелей) і чинників середовища. Цю групу хвороб називають хворобами із спадковою схильністю.
Причини та особливості розвитку даних хвороб досить складні, багаторівневі, остаточно не з’ясовані. Звичайно вони різні для кожної хвороби. Проте стосовно спільних особливостей розвитку таких хвороб існує загальновизнане уявлення.
В основі спадкової схильності до хвороб лежить широкий генетично збалансований поліморфізм популяцій людини за ферментами, структурними і транспортними білками, а також за антигенними системами. У популяціях людини принаймні 25-30% локусів (із близько 40 000) представлено двома і більше алелями. Отже, індивідуальні комбінації алелей неймовірно різноманітні. Вони забезпечують генетичну унікальність кожної людини, яка виражається не тільки в здібностях, фізичних відмінностях, але і в реакціях організму на патогенні чинники навколишнього середовища. Хвороби із спадковою схильністю виникають у осіб з відповідним генотипом (поєднання «приваблюючих» алелей) при провокуючій дії чинників середовища.
Хвороби із спадковою схильністю умовно можна розділити на такі основні групи: 1) природжені вади розвитку; 2) поширені психічні та нервові хвороби; 3) поширені хвороби середнього віку.
Найпоширенішими вродженими вадами розвитку є розщеплення верхньої губи та піднебіння, вивих стегна, клишоногість тощо. До поширених психічних і нервових хвороб із спадковою схильністю належать шизофренія, епілепсія, маніакально-депресивні психози, розсіяний склероз та інші. Серед соматичних хвороб середнього віку відомі псоріаз, бронхіальна астма, виразка шлунку та 12-палої кишки, ішемічна хвороба серця, гіпертензія, цукровий діабет тощо.
Механізми виникнення хвороб із спадковою схильністю, не дивлячись на їх складність, все більше і більше піддаються генетичному аналізу у зв’язку з успіхами розшифрування генома людини. У патогенезі цих хвороб переважне значення мають спадкові чинники. Патогенез такої хвороби – складний, багатогранний і багаторівневий процес, тому значення спадкових чинників неможливо визначити однозначно у всіх випадках. Часто важко відділити чинники один від одного як стосовно інтенсивності, так і тривалості їх дії. Розуміння причин і перебігу хвороб із спадковою схильністю ускладнюється ще й тим, що їх розвиток – результат взаємодії генетичних факторів (моногенних чи полігенних) з чинниками середовища, іноді дуже специфічними, іноді менш специфічними. Лише найновіші досягнення у вивченні геному та складанні генних карт хромосом людини дозволяють наблизитися до виявлення ефектів головного аномального гена.
Насправді кожне захворювання із спадковою схильністю – це генетично гетерогенна група з однаковим клінічним кінцевим проявом. У кожній групі є декілька різновидів, які обумовлені різними генетичними та негенетичними причинами. Наприклад, групу ішемічних хвороб серця можна розділити на кілька моногенних форм гіперхолестеринемій (підвищений вміст холестерину в крові).
Причини розвитку хвороб із спадковою схильністю представлені на мал. 55. Кількісне поєднання кожної з цих причин у розвитку захворювань може бути різним у різних людей.
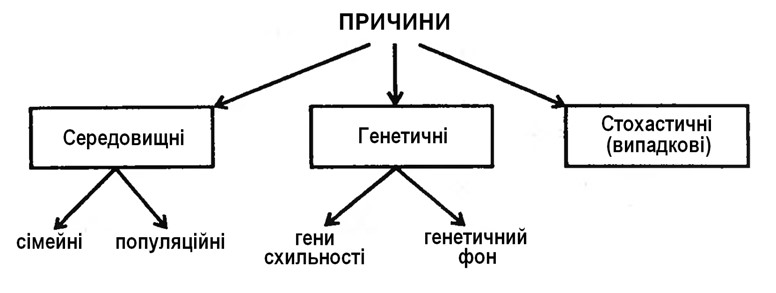
Мал. 55. Причини хвороб із спадковою схильністю
Для прояву хвороб із спадковою схильністю необхідне конкретне поєднання спадкових і зовнішніх чинників. Чим більше виражена спадкова схильність і більше шкідливих факторів середовища, тим для індивіда вище вірогідність захворіти (і в більш ранньому віці, і у важчій формі). Порівняльне значення зовнішніх та спадкових чинників у розвитку хвороб схематично показано на мал. 56.
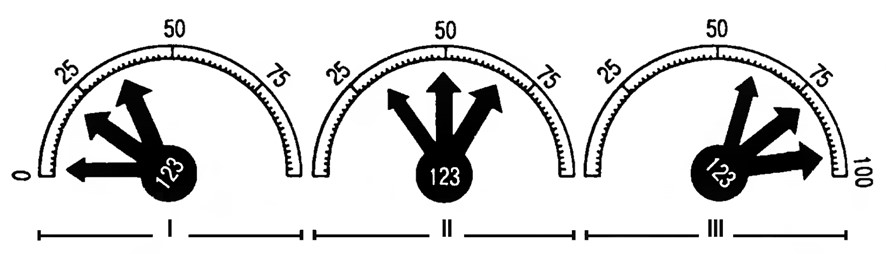
Мал. 56. Співвідношення ролі генетичних факторів та чинників оточуючого середовища у розвитку хвороб із спадковою схильністю:
I, II, III – ступені спадкової схильності (слабка, помірна, сильна відповідно); 1,2,3 – провокуючі чинники середовища різної сили; по шкалі – частка хворих (%).
На схемі умовно визначено три рівня спадкової схильності та три ступені дій середовища: слабкий, помірний та сильний. При слабкій спадковій схильності та незначному впливові середовища організм підтримує гомеостаз і хвороба не розвивається. Однак при посиленні дії шкідливих чинників середовища у певної частини осіб виявиться хвороба. За умови значної спадкової схильності до патології одні й ті ж чинники середовища викликають нездужання у більшої кількості людей.
Хвороби із спадковою схильністю відрізняються від інших форм спадкової патології (генних і хромосомних хвороб) характером клінічної картини. На відміну від генних хвороб, при яких всіх членів сім’ї пробанда можна розділити на хворих і здорових, клінічна картина хвороб із спадковою схильністю має безперервні переходи в межах однієї і тієї ж форми патології.
Для хвороб із спадковою схильністю характерна відмінність в їх прояві і тяжкості перебігу залежно від статі та віку. Механізми поширеності таких хвороб за часом достатньо складні, оскільки в популяціях як генетичні характеристики схильності, так і чинники середовища можуть змінюватися в той чи інший бік.
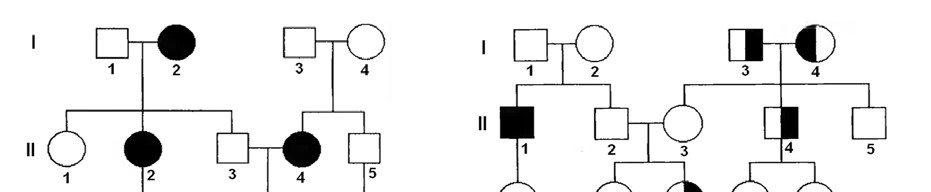
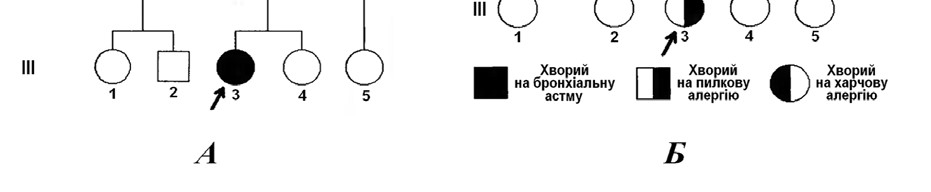
Мал. 57. Родоводи з гіпертензією (А) та алергічними патологіями (Б)
Одна з характерних особливостей хвороб із спадковою схильністю – підвищена частота (накопичення) в певних сім’ях, яка обумовлена їх генетичною конституцією. На мал. 57 приведені приклади родоводів, «обтяжених» гіпертензією та алергічними хворобами. Генеалогічний аналіз таких родоводів дає змогу краще визначити прогноз перебігу патології в сім’ї, а також лікувальні та профілактичні заходи проти неї.
Спадкова схильність до хвороби може мати моногенну або полігенну основу.
5.7.1. Моногенно зумовлена схильність до хвороб
Моногенно обумовлені форми спадкової схильності до хвороб спричинюються мутаціями окремих генів. Ця схильність, як правило, успадковується за аутосомно-рецесивним або Х-зчепленим рецесивним типом. Проте розщеплення за патологічною ознакою в поколіннях не відповідає менделевим законам, оскільки носій аномального гена протягом життя контактує з провокуючими хворобу чинниками середовища, які можуть мати мінливий характер.
Нині виявлено понад 40 генів, які беруть участь у біотрансформації сторонніх або шкідливих речовин, що надходять в організм із оточуючого середовища. У нормі вони продукують такі ферменти як цитохром Р450, Nацетилтрансфераза, пароксоназа сироватки, холінестераза сироватки, глюкозо-6-фосфатдегідрогеназа, лактаза, інгібітори протеаз тощо. Патологічні реакції організму на сторонні речовини викликаються мутантними алелями цих генів. При цьому кожному гену відповідає конкретний специфічний провокуючий чинник. Варіабельність «концентрації» таких чинників (найчастіше хімічних речовин у складі їжі, води, повітря) призводить до того, що одна й та ж хвороба виявляється по-різному навіть у межах однієї сім’ї.
Джерелом провокуючих факторів можуть бути забруднена атмосфера, продукти харчування та харчові добавки, фізичні фактори, біологічні агенти тощо.
Забруднення атмосфери вихлопними газами, газоподібними речовинами численних виробничих об’єктів є серйозною екологічною проблемою. З деякими з цих шкідливих чинників людина стикається завжди, з іншими – зрідка. Хімічні сполуки та пилові частинки з повітря, потрапляючи в організм через легені, шкіру і слизові оболонки, можуть викликати певні реакції спадкового характеру.
Найбільш вивченою мутацією, що обумовлює реакцію на забруднення атмосфери, є недостатність α1-антитрипсина (білка сироватки крові), який є інгібітором (стримувачем) протеаз (ферментів, що розщеплюють білки та пептиди). Цей білок продукується геном РІ, який розташований в довгому плечі 14-ої хромосоми (14q32). В нормі концентрація α1-антитрипсина підвищується при різних фізіологічних і патологічних станах (вагітність, запалення тощо). Різні його форми (продукти алелей PIM, PIS, PIZ) розрізняються за антитрипсиновою активністю. Неактивна форма білка спричинюється алелем PIZ і є рецесивною ознакою. Частота гомозигот PIZPIZ у європейців складає 0,05%, гетерозигот – 4,5%.
Особи із спадковою недостатністю інгібітору протеаз, якщо вони гомозиготні за мутантним алелем (PIZPIZ), у віці 30-40 років надзвичайно схильні до розвитку хронічних запальних захворювань і емфіземи легенів. Остання у таких осіб розвивається в 30 разів частіше, ніж у популяції, і протікає дуже важко. Куріння та запилене повітря істотно прискорюють розвиток емфіземи. Навіть у осіб, гетерозиготних за геном недостатності інгібітору протеаз (генотип PIМPIZ), частота яких в деяких популяціях перевищує 10%, наявні патологічні реакції на підвищену запиленість повітря та куріння, тобто підвищений ризик розвитку емфіземи легенів. Отже, для цієї групи осіб необхідно виключити вплив виробничих пилових чинників, щоб запобігти розвитку хвороби. Методи визначення недостатності α1-антитрипсина розроблені і можуть застосовуватися при професійних оглядах і відборах для роботи на відповідних підприємствах.
При наявності у вдихуваному повітрі вуглеводнів (куріння та відповідні виробництва) особи, що відзначаються високим вмістом арилгідрокарбонгідроксилази в організмі (домінантна гомозигота за алелем Val гена CYP1A1; локалізація 5q22- q24), мають досить значний ризик захворіти на рак легенів.
Знайдена також генетична схильність до раку сечового міхура. Вона пов’язана з мутаціями в локусі N-ацетилтрансферази печінки (ген NAT2; локалізація 8p23.1- р21.3). Під дією цього ферменту канцерогенні речовини розкладаються та виводяться з організму. За швидкістю знешкодження канцерогенів розрізняють три фенотипи: швидкі (гомозиготи за нормальним алелем), повільні (гомозиготи за мутантним алелем) та проміжні (гетерозиготи). Рак сечового міхура частіше розвивається у повільних фенотипів. Ризик особливо підвищується при дії чинників середовища (куріння, виробництво гумових виробів, фарби).
Продукти харчування. Певні харчові продукти можуть викликати небажані реакції у генетично чутливих індивідів.
Одним із відомих захворювань цієї групи є непереносимість лактози (молочний цукор). У осіб з цим дефектом після вживання молока виникає розлад травлення. Суть дефекту зводиться до відсутності в кишечнику синтезу лактази (фермент, що розщеплює лактозу), внаслідок чого нерозщеплена лактоза служить сприятливим субстратом для розмноження гнильної мікрофлори. Мутантні алелі R гена лактази (ген LАC, мутантний алель позначається LАC*R; локалізація 2q21) досить поширені у східних народів (до 95– 100%) та у американських індіанців і афроамериканців (70–75%). Серед європейців частота гомозигот за цими мутаціями невелика (5–10%).
Іншими прикладами захворювань цієї групи є целіакія (непереносимість білків злаків), спадкова недостатність глюкозо-6-фосфатдегідрогенази (непереносимість зерен кінських бобів, деяких ліків, промислових окислювачів; ген G6PD; локалізація Xq28), гемікранія, або мігрень (непереносимість твердих сирів та шоколаду), відсутність одного з двох генів альдегіддегідрогенази печінки (токсична реакція на алкоголь; ген ALDH2; локалізація 12q24.2).
Фізичні чинники та метали. Характерним прикладом спадкової чутливості до фізичних чинників (УФ промені) є одна із форм пігментної ксеродерми (див. розділ 5.4.3.), яка успадковується за аутосомно-рецесивним типом.
У науковій літературі описана також різна чутливість до солей важких металів (свинець, ртуть, кадмій тощо). Наприклад, отруєння органічними сполуками ртуті викликає нейропсихічні розлади різного рівня у різних осіб. Гетерозиготні носії генів цистинозу та анемії Фанконі можуть бути схильні до токсичної дії металів або інших ниркових токсинів. Підвищена, хоча і не «токсична» присутність свинцю може бути провокуючим чинником гіперактивної поведінки у дітей із спадковою схильністю.
Біологічні агенти. Імунітет визначається генетичними механізмами і має неоднаковий рівень прояву у різних особин. Добре відомі факти різної чутливості людей до вакцин при введенні одних і тих же доз препаратів. У деяких людей з’являються чіткі клінічні прояви хвороби, у інших реакція на імунізацію абсолютно відсутня. Це є наслідком генетичного поліморфізму реакцій на дію зовнішніх біологічних чинників.
Класичним прикладом спадкової стійкості до біологічних агентів служать деякі спадкові хвороби, наприклад, серпоподібно-клітинна анемія (мутація гена HBB; локалізація 11p15.5) та недостатність глюкозо-6фосфатдегідрогенази (див. вище). У цих випадках малярійний плазмодій не може розмножуватися в еритроцитах мутантних гомозигот і гетерозигот. Характерно, що в регіонах з високою захворюваністю на малярію (Африка, Греція, Італія, Філіппіни, Азербайджан, Узбекистан) спостерігається висока частота мутантних алелей генів, які спричинюють серпоподібно-клітинну анемію та недостатність глкжозо-6-фосфатдегидрогенази.
5.7.2. Полігенно зумовлена схильність до хвороб
Полігенна спадкова схильність до хвороб визначається поєднанням алелей декількох генів. Кожний алель окремо скоріше нормальний, ніж патологічний, а спричинює хворобу певна їх комбінація. Ідентифікація цих генів та їх алелей вельми складна. Свій патологічний потенціал вони проявляють разом з комплексом декількох чинників оточуючого середовища. Патології, спричинені такими ситуаціями, дістали назву мультифакторних хвороб. Співвідношення значень генетичних і середовищних чинників різна не лише для даної хвороби, але і для кожного хворого.
Для визначення полігенної спадкової схильності до хвороб застосовують, як і у випадку моногенної схильності, три основні методи – генеалогічний, близнюковий та популяційно-статистичний. При цьому кожний метод має певні обмеження (порівняно з вивченням моногенних форм), які слід враховувати при проведенні дослідження. Слід також підкреслити, що для проведення таких досліджень потрібно набагато більше родоводів чи близнюкових пар, ніж при вивченні моногенних ознак. Інколи для вирішення однієї задачі необхідно проаналізувати декілька сотень і навіть тисяч родоводів.
Полігенні патології відзначаються такими характерними властивостями:
- чим рідше зустрічається хвороба в популяції, тим вище ризик для родичів пробанда і тим більше різниця у величині ризику між родичами 1–2 та 2–3 ступенів спорідненості;
- чим сильніше виражена хвороба у пробанда, тим вище ризик захворювання для його родичів;
- ризик для родичів пробанда буде вище, якщо є іншій хворий кровний родич;
- якщо частота хвороби варіює залежно від статі, то ризик для родичів буде вищий, якщо пробанд відноситься до статі, що менше уражена.
Мультифакторні хвороби, як і будь-які інші подібні ознаки, можуть бути непереривними та переривистими.
Неперервні мультифакторні ознаки. Велика кількість нормальних і патологічних ознак людини є неперервними мультифакторними ознаками. Для них характерний поступовий, плавний перехід від мінімальних до максимальних їх значень. Такий розподіл ознаки називається нормальним. При цьому більшість особин знаходиться поблизу її середнього значення. (Мал. 58).
Крім морфологічних характеристик людини (див. розділ 2.2.), нормальний розподіл властивий також таким ознакам як артеріальний тиск крові, кількість лейкоцитів у крові, температура тіла, емоційність, пам’ять, інтелект та багато інших.
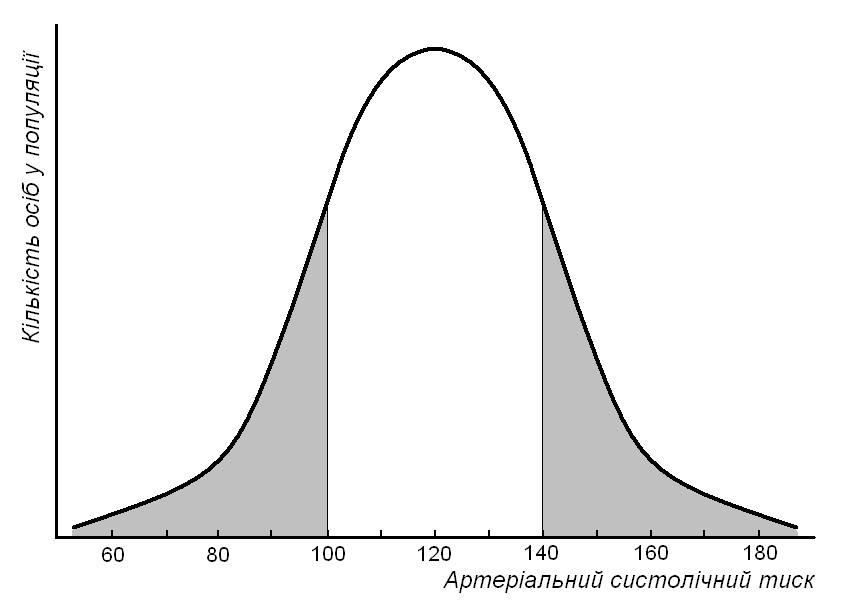
Мал. 58. Крива розподілу значень артеріального тиску крові в популяції людини
Середнє значення таких ознак вважається нормальним, а зменшення чи збільшення їх значення – аномальним. Так, нормальний артеріальний тиск крові (в мм ртутного стовпчика) знаходиться в межах 100-140 (систолічний тиск) та 60-90 (діастолічний тиск) (мал. 58). Якщо людина має кров’яний тиск, нижчий цих показників, то вона страждає на гіпотензію, а якщо вищий – то на гіпертензію. У обох випадках – це досить серйозні патології. Гіпотензія супроводжується головним болем, слабкістю, іноді – втратою свідомості. Перебіг гіпертензії при відсутності ускладнень звичайно безсимптомний і виявляється лише при вимірюванні тиску. Можливі симптоми цієї аномалії – періодичний головний біль, запаморочення, миготіння «мух» перед очима, шум у вухах, біль у зоні серця, серцебиття. З часом гіпертензія спричинює патологічні зміни кровоносних судин, порушення функцій серця, мозку, нирок, зору. Розповсюдженість гіпертензії серед людей старшого віку складає 10-20%.
Переривисті мультифакторні ознаки. Найбільш відомими переривистими мультифакторними патологіями є такі вроджені вади розвитку як клишоногість, вивих стегна, звуження воротаря шлунку, розщеплення верхньої губи та піднебіння і такі поширені захворювання дорослих як аномалії серця, ревматоїдний артрит, виразкова хвороба, бронхіальна астма, цукровий діабет, шизофренія, епілепсія та інші.
Особливості успадкування таких патологій добре демонструються на прикладі вродженої вади розвитку – розщеплення верхньої губи та піднебіння (мал. 59). Батьки дитини з даною вадою, як правило, здорові. Проте народження хворої дитини свідчить, що кожний з них є носієм багатьох умовно аномальних алелей, кількість яких все ж таки недостатня, щоб дефект сформувався у батьків. Якщо дитина випадково успадкує критичну кількість «аномальних» алелей, тобто певна межа буде перевищена, виникає вада розвитку – розщеплення верхньої губи та піднебіння.
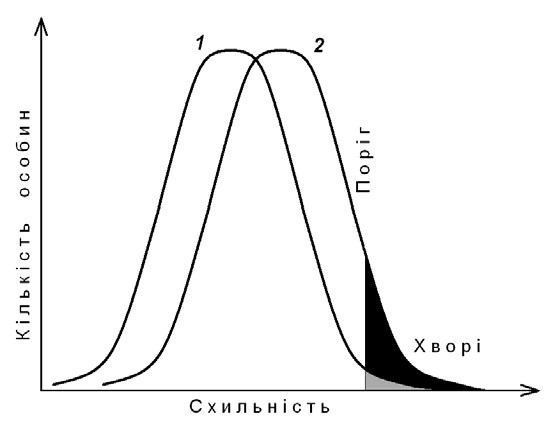
Мал. 59. Розподіл схильності до розщеплення верхньої губи та піднебіння в популяції (1) та в родичів пробанда (2)
Схильність до цієї патології визначається як генотипом, так і факторами середовища і характеризується кривою нормального розподілу. Частина популяції, розташована праворуч від межового рівня, показує частоту захворювання в популяції – 0,1%. Однак, для батьків хворої дитини крива схильності зсовується вправо. Це означає, що для родичів першого ступеня спорідненості частота (або ризик) захворювання складає вже 4 %. Близькість до патологічної межі конкретних індивідів у популяції відзначається накопиченням у них певних незначних аномалій (розщеплення язичка, дефекти зубів і прикусу, асиметрія прикріплення крил носа), які знаходяться в зоні розвитку вади.
Якщо серед особин лівої частини популяції хворі зустрічаються рідко, то по мірі руху вправо частота хворих збільшується аж до переважної більшості в крайній правій частині кривої. Це означає, що при низькій генетичній схильності до патології необхідне дуже несприятливе поєднання багатьох чинників оточуючого середовища, щоб хвороба проявилася. Однак за високої генетичної схильності захворювання здатне розвинутися і без видимих провокуючих факторів середовища.
5.7.3. Генетика онкологічних патологій
Онкологічні хвороби займають одне з перших місць серед поширених мультифакторних спадкових патологій як за різноманітністю форм, так і за популяційною частотою. Генеалогічний та близнюковий методи дослідження виявили значну роль генетичних чинників у розвитку онкопатологій. Однак було також доведено, що не менше значення у цьому процесі мають фактори оточуючого середовища. Взяти хоча б відомі професійні форми раку у рентгенологів, рак шкіри у осіб, що контактують з кам’яновугільною смолою, рак сечового міхура у працівників анілінових виробництв, первинний рак печінки як наслідок вірусного гепатиту, рак легенів у працівників азбестових заводів тощо.
Попри великі успіхи в молекулярній генетиці, залишаються поки-що невідомими численні процеси взаємодії генетичних і середовищних чинників у розвитку злоякісних новоутворень.
Злоякісні новоутворення є генетичними хворобами соматичних клітин. Механізми генетичних соматичних хвороб дуже складні, внаслідок чого їх важко класифікувати. Мутації, що визначають розвиток пухлини, можуть трапитися в статевих або соматичних клітинах. У першому випадку вони існували вже в гаметі і, отже, присутні у всіх клітинах організму, в другому – виникають у соматичній клітині як результат постійного мимовільного або спровокованого мутаційного процесу. Виникнення пухлини часто починається в соматичних клітинах з мутації того ж локуса, в якому вже є мутація гаметного походження.
Звичайно неможливо чітко розмежувати етапи злоякісних новоутворень людини, обумовлені спадковістю та викликані чинниками середовища. Однак у окремих випадках це вдається зробити за допомогою ряду методів дослідження – популяційно-статистичного, генеалогічного, близнюкового, генетичних маркерів, біохімічного та в дослідах на тваринах.
Популяційні дослідження показують, що поширеність таких форм злоякісних новоутворень, як рак молочної залози та рак шлунку, в різних популяціях розрізняється. Наприклад, частота раку молочної залози у жінок Західної Європи та Північної Америки у 8 разів вище, ніж у японок чи китаянок, а ризик виникнення раку шлунку у японців і китайців у 10 разів нижче, ніж у європейців. Проте різниця зменшується, хоча і не ліквідовується, через 2-3 покоління за умови життя китайців і японців у країнах Західної Європи та Північної Америки.
Вивчення родоводів показують, що якщо жінка хвора на рак молочної залози, то у її родичок I ступеня спорідненості ризик виникнення тієї ж форми раку зростає в 2-3 рази. Подібні результати одержані також у випадку раку шлунку.
Спадкова схильність до злоякісних новоутворень у меншій мірі виявляється в близнюкових дослідженнях. Тут теж наявна різниця в рівні конкордантності монозиготних та дизиготних близнюків, але вона незначна.
Провокуючими факторами розвитку злоякісних пухлин є окремі форми вірусів, які інтегруються в геном клітин. Вивчення їх ролі привело до відкриття так званих онкогенів у клітинах ссавців. У нормальних клітинах цих тварин може бути два типи послідовностей ДНК, гомологічних вірусним онкогенам. Один із типів назвали протоонкогенами, які фактично є нормальними генами. Інший тип дістав назву клітинних онкогенів, тобто генів з онкогенними властивостями. На сьогодні виявлено понад 100 онкогенів: ras, myc, fos, mos та інші.
У нормальному стані протоонкоген бере участь у контролі клітинного циклу, а при підвищенні його експресії перетворюється на клітинний онкоген, провокуючи розвиток злоякісної пухлини одним із чотирьох способів.
- Внаслідок інтеграції ДНК, комплементарної РНК ретровіруса (так званої кДНК), в хромосому поблизу онкогена може розпочатися його неконтрольована експресія.
- Збільшення числа копій онкогена в сотні разів веде до значного зростання концентрації онкопротеїнів у клітині. Такий механізм характерний для певних пухлин, наприклад, дрібноклітинного рака легень, до якого призводить неконтрольоване копіювання онкогенів C-myc, N-myc та L-myc.
- Можуть трапитися мутації протоонкогенів, які спричинять їх активацію та накопичення онкогенних білків. Може також спостерігатися неконтрольоване копіювання мутантного онкогена, що посилює онкогенні властивості клітини.
- Хромосомні транслокації можуть порушувати біохімічну функцію або рівень активності протоонкогена через інше його «оточення». Прикладом цього може бути хронічний мієлоїдний лейкоз, при якому ділянка довгого плеча хромосоми 22 переміщена на довге плече хромосоми 9, а зовсім невелика частина хромосоми 9 відповідно приєднана до хромосоми 22. В результаті цієї транслокації, яка позначається t(9;22)(q34; q11), клітинний онкоген C-abl з хромосоми 9 переноситься в регіон гена bcr хромосоми 22, що приводить до синтезу химерного продукту, що має онкогенні властивості.
В експериментах з гібридизації злоякісних та нормальних клітин були виявлені особливі аутосомно-домінантні гени, які дістали назву антионкогенів – генів-супресорів (генів-пригнічувачів) онкогенів. Сьогодні відомо понад 40 таких генів (Rb, Wtl, BRCA1, BRCA2 тощо), але припускається, що кількість їх у нормальній клітини відповідає кількості онкогенів. Антионкогени пригнічують неконтрольований поділ клітин. Якщо обидва алелі антионкогена (один – батьківського походження, а інший – материнського) виявляться мутантними або втраченими внаслідок делеції, то в такому організмові починається неконтрольований поділ клітин. На даний час виявлені певні закономірності дії генів-супресорів пухлин.
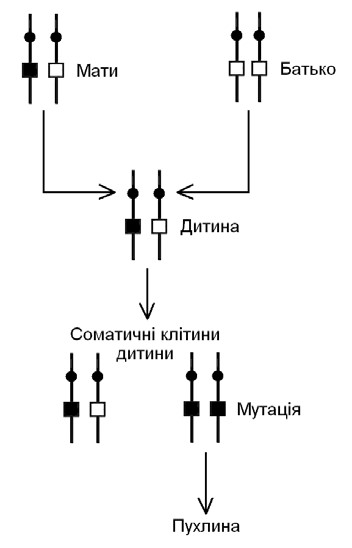
Мал. 60. Втрата гетерозиготності по гену-супресору пухлини: □ – нормальний алель гена-супресора, ■ – аномальний алель гена-супресора
Людина може успадкувати від одного з батьків аномальний алель генасупресора. Це може бути мутація або мікроделеція цього гена. Гетерозиготність індивіда за даним локусом убезпечує його від виникнення пухлини, оскільки ген-супресор є домінантним. Проте в процесі життя в соматичних клітинах постійно відбувається мутаційний процес, який може торкнутися і нормального алеля антионкогена. При цьому клітина стає гомозиготною за аномальним геном, внаслідок чого припиняється пригнічення онкогена і розвивається злоякісна пухлина. (Мал. 60).
У людини виявлено та описано досить значну кількість онкопатологій, спричинених втратою гетерозиготності по гену-супресору пухлин. Для більшості з них відомі не тільки локалізація гена, але і його структура, первинні продукти та можливі мутації. Цікаво, що для виникнення певних злоякісних пухлини необхідна втрата гетерозиготності не в одному, а в кількох локусах.
Крім того, потрібні ще мутації в самих онкогенах. Прикладами можуть бути рак нирки (локалізація 3р), рак легень (3р, 13q, 17p), рак молочної залози (1q, 3р, 13q, 17р), рак печінки (11р), рак товстої кишки (5q, 17р, 18q) та інші. Це свідчить про полікомпонентність генетичного механізму розвитку онкопатології.
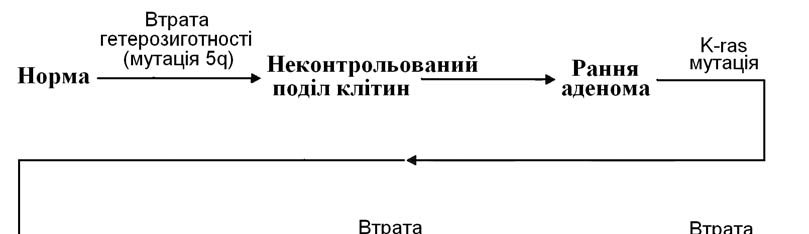
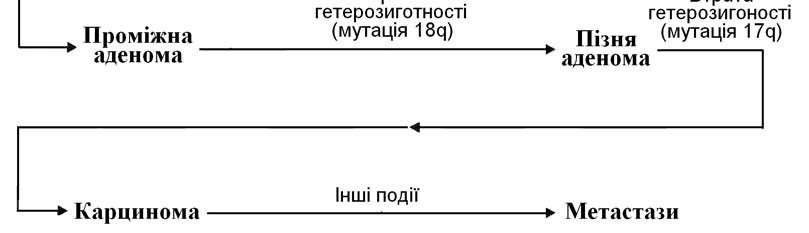
Мал. 61. Роль генетичних процесів у розвиткові раку товстої кишки
На мал. 61 показано роль генетичних подій у патогенезі раку товстої кишки. До них відносяться гетерозиготність за локусом 5q (норма), втрата цієї гетерозиготності, мутація онкогена K-ras і т.д. Перетворення доброякісної пухлини (аденоми) в злоякісну (карциному) відбувається за рахунок накопичення мутацій та втрати гетерозиготності. Послідовність генетичних подій не має принципового значення для кінцевого ефекту (пухлини). Важливо, щоб у клітині відбулися всі події. Наприклад, при втраті гетерозиготності 18q і 17q до того, як виникне мутація К-ras, не може початися формування аденоми. Проте, як тільки відбудеться мутація в гені К-ras, стадія аденоми пройде дуже швидко, оскільки в тканині вже є передумови для інтенсивного розмноження клітин з причини втрати гетерозиготності 18q та 17q.
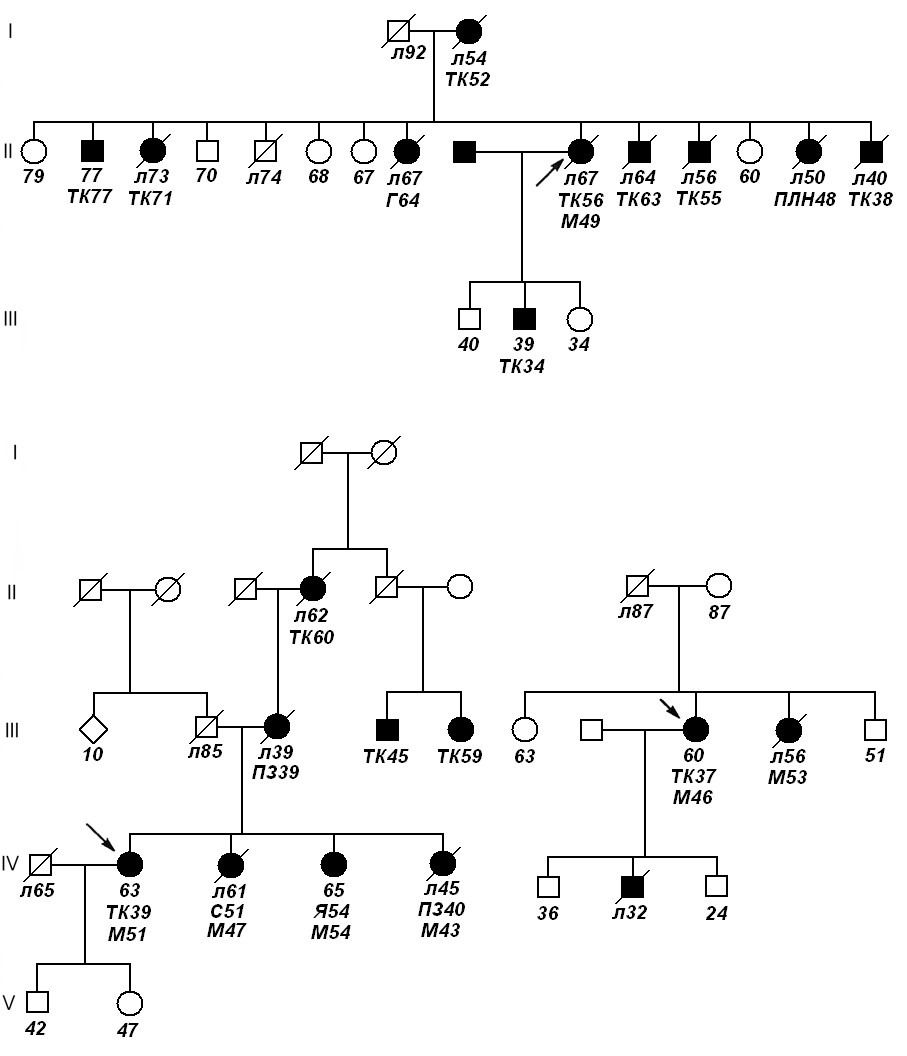
Мал. 62. Родоводи трьох онкологічних сімей:
ТК – рак товстої кишки, ПЗ – рак підшлункової залози, С – рак сечівника, Я – рак яєчника, Г – рак горла, ПЛН – початкова локалізація пухлини невідома, М – рак матки, л – смерть; числа показують вік виявлення патології, смерті або обстеження.
Разом з формами злоякісних новоутворень, що розвиваються на основі успадкованих мутацій (це близько 5% випадків раку молочної залози та товстої кишки), є також сімейні синдроми, що схиляють до раку. Є ціла низка ознак наявності таких синдромів у родоводі: 1) широке розповсюдження злоякісних пухлин у родичів І та II ступенів спорідненості, 2) випадки схожих форм раку у близьких родичів (наприклад, молочної залози та яєчника, кишечника та матки), 3) наявність двох членів сім’ї зі схожими рідкісними формами раку, 4) незвичайно ранній вік розвитку онкопатології, 5) двосторонні пухлини парних органів, 6) синхронність або безперервність виникнення пухлин, 7) пухлини в органах двох різних систем у одного індивіда. Нижче (мал. 62) приведені три родоводи, обтяжені злоякісними новоутвореннями.
Описано вже близько двох десятків спадкових синдромів, які виявляють сімейну схильність до онкопатологій. Серед них – неполіпозний рак товстої кишки, аденоматозний поліпоз, ретинобластома, рак молочної залози та інші. Для всіх цих захворювань відома локалізація гена, а для деяких і його структура, що дозволяє проводити їх генетичну діагностику задовго до розвитку пухлин.
Розглянуті вище складові канцерогенезу (протоонкогени, клітинні онкогени, антионкогени, втрата гетерозиготності за генами-супресорами, зчеплення з генетичними маркерами) не вичерпують всієї різноманітності компонентів генетичної схильності до раку та причин пухлинного процесу. Можна визначити щонайменше ще дві групи спадкових характеристик індивіда, що стосуються канцерогенезу, – особливості репаративних (відновлювальних) процесів та біохімічних систем в організмі.
У підтримці динамічної стабільності генетичних структур клітини, які визначають нормальну поведінку клітини, істотну роль відіграють репаративні процеси. Організм людини має природну здатність до репарації пошкоджень ДНК (мутацій генів), які виникають спонтанно або під впливом сторонніх чинників. Спадкові аномалії в системах репарації ДНК ведуть до злоякісних новоутворень – пігментної ксеродерми, спадкового неполіпозного раку товстої кишки тощо.
Біохімічні системи організму людини надзвичайно складні та розгалужені. Окремі з них представлені генетично поліморфними формами. Різноманітність ферментів, які активізують (естерази, оксигенази, цитохром Р450) або знешкоджують (різноманітні трансферази) сторонні для організму сполуки, визначає індивідуальну чутливість до канцерогенних факторів.
Розглянуті вище особливості розвитку та перебігу онкологічних захворювань свідчать про те, що аналіз родоводів та надання медико-генетичної консультації з приводу онкологічних патологій є надзвичайно складною справою.