Генетика людини: методичка
6.2. Розумова відсталість
Розумовою відсталістю, чи інтелектуальною недостатністю називають стійке порушення пізнавальної діяльності внаслідок ураження головного мозку. Ця вада досить неоднорідна за багатьма показниками – за причинами, клінічною картиною, динамікою, психологічною структурою порушення тощо. Нині розумова відсталість вважається однією із досить поширених вад розвитку. Її популяційна частота у різних країнах світу складає 0,4-2,7 %. До того ж вона має тенденцію до зростання.
Близько 80% розумово відсталих дітей страждає на олігофренію. Більшість олігофренів має значення коефіцієнта інтелекту в межах 51-70 балів. Це так звані дебіли. Більш тяжкі порушення інтелекту (21-50 балів) у імбецилів. При значенні коефіцієнта інтелекту в межах 0-20 балів наявна ідіотія.
Олігофренія характеризується цілим комплексом патологічних ознак:
- затримка загального розвитку;
- недостатність абстрактного мислення;
- еволютивний (повільний) характер динаміки хвороби;
- ранній (до 2-2,5 років – домовний період) початок патологічних змін.
Набута недоумкуватість (деменція) виникає пізніше, ніж олігофренія. Вона може бути викликана найрізноманітнішими причинами: зовнішнє органічне ураження головного мозку, несприятливий перебіг шизофренії та епілепсії, деякі спадкові дегенеративні хвороби.
У клінічній картині деменції (на відміну від олігофренії) переважають явища порушення вже сформованих (хоча б частково) функцій. Якщо ознаки затримки розвитку і наявні, то вони мають другорядне значення. Віддалений прогноз динаміки деменції, що особливо важливо для корекційної педагогіки, може бути самими різним – від еволютивного до чітко прогредієнтного (див. розділ 5.3.3.).
Однозначне розмежування деменції та олігофренії можливе не завжди. Особливо це важко визначити у віці 2-4 років, коли ознаки затримки розвитку та пошкодження цілком співвідносні за своїми внесками в структуру клінічних проявів. Проте розмежування цих патологічних явищ бажане навіть з чисто практичних позицій, оскільки кожний з цих видів розумової відсталості вимагає особливої стратегії корекційних заходів і може розрізнятися за прогнозом. Тут доречно зауважити, що 3/4 випадків розумової відсталості є генетично обумовленими і лише 1/4 викликана зовнішніми чинниками.
Слід підкреслити, що розумова відсталість при спадкових хворобах є, як правило, одним з симптомів у складній клінічній картині захворювання. Для деяких спадкових хвороб вона є обов’язковим симптомом, а для інших – фігурує не у всіх випадках.
6.2.1. Розумова відсталість при порушення кількості хромосом
На частку олігофренії, обумовленої різними хромосомними порушеннями, припадає близько 10-12 % усіх випадків розумової відсталості.
Головна клінічна особливість хромосомних хвороб – розумова відсталість і множинні вади розвитку. Серед дітей з множинними вадами розвитку хромосомні хвороби складають близько 43 % випадків.
Серед хвороб, викликаних кількісними порушеннями аутосом, найпоширенішим є синдром Дауна (див. розділ 5.5.2.). Усі хворі на синдром Дауна характеризуються інтелектуальною недостатністю різного рівня: 5 % – легкою, 75 % – помірною та вираженою, 20 % – важкою.
Хворі діти мають мляве мислення, не здатні до абстракції, через силу опановують прості арифметичні дії. У часові вони орієнтуються гірше, ніж у просторі. Читання дається легше, ніж письмо – більшість читає нашвидку і навіть виразно, копіюючи модуляції голосу дорослих. Переказувати прочитане діти з синдромом Дауна здатні тільки з питаннями; самостійний переказ дається з великими труднощами або неможливий.
Мова розвивається пізно: перші слова за типової хвороби з’являються на п’ятий рік, а прості фрази – на восьмий. Особливо слабкою є активна спонтанна мовна діяльність. Запас слів бідний, з багатьох причин (недорозвинена верхня щелепа, патологія зубних рядів, надто великий язик) порушене звукоутворення.
Емоції у хворих на синдромом Дауна бідні, діти несамостійні, не проявляють ініціативи, надто піддаються навіюванням, схильні до наслідування, імпульсивні (поривні, непередбачувані), невмотивовано вперті, часто проявляють негативізм (особливо до всього нового). Однак, емоційна сфера у них, порівняно з інтелектом, проявляється краще. Діти з синдромом Дауна добре розрізняють ставлення до себе оточуючих, багато хто з них чуйний та посвоєму дбайливий, їм властиве відчуття сорому, образи, збентеження, співпереживання.
У хворих на синдром Дауна спостерігається різноманітний темперамент. Одні з них метушливі, неспокійні, дуже допитливі, в усе втручаються, товариські, але полохливі, багато чого бояться, перш за все новизни. У відносинах з іншими часто ласкаві, охоче розмовляють, спроможні користуватися жестами. Радісно сприймають похвалу, ревниві, вимагають до себе уваги і не люблять, щоб її спрямовували на інших. Разом з тим ці діти дратівливі та злі, здатні нишком образити слабшого. Однак у більшості випадків вони дружні та доброзичливі.
Інші діти малорухливі, незграбні, замкнуті, байдужі, при спробі залучити в розмову відповідають односкладово і часто невлад («забув», «не знаю», «не пам’ятаю» тощо). В роботу включаються поволі, але якщо це відбувається, працюють вельми старанно і терпляче. Емоційні особливості таких дітей можуть бути самими різними.
Синдрому Дауна приділяється велика увага не тільки тому, що він зустрічається частіше, ніж інші хромосомні порушення. Це приклад того, як знання клінічних особливостей (схильність до наслідування, цікавість, у деяких – посидючість, значне збереження емоційної сфери тощо) дозволяє вибрати опорні позиції для побудови навчально-корекційного процесу. Ці знання також дають змогу прогнозувати поведінку учня, особливо в період статевого дозрівання, який характеризується проявами дратівливості, грубості, підвищеної сексуальності, нестійкого настрою в поєднанні з деяким зростанням інтелектуального потенціалу.
При порушенні числа статевих хромосом розумові та психічні аномалії відрізняються меншою тяжкістю, ніж при порушеннях числа аутосом. У цих випадках часто відсутня або виявляється в легких формах розумова відсталість; зате порушення емоційно-вольової сфери звичайно досить виразні і нерідко відносяться до провідних діагностичних ознак захворювання.
Прикладами хвороб, спричинених порушеннями кількості статевих хромосом, є синдроми Шерешевського – Тернера та Клайнфельтера (див. розділ 5.5.3).
При синдромі Шерешевського – Тернера (хворіють лише жінки) відставання розумового розвитку незначне і зустрічається лише у 16-25 % випадків. Решта хворих відзначається вузькістю інтересів, низьким потенціалом мислення, підлеглістю та своєрідним життєвим практицизмом.
Дівчатка з синдромом Шерешевського – Тернера добрі, працелюбні, нерідко схильні до повчання, люблять опікувати молодших, поратися вдома. При настанні статевої зрілості нерідко виникають невротичні реакції, пов’язані з усвідомленням своєї неповноцінності. Хворі стають замкнутими, дратівливими, часто ведуть себе брутально.
Синдром Клайнфельтера зустрічається тільки у хлопчиків. При цьому розумова відсталість виявляється у 25-50 % випадків і варіює від примежних станів до дебільності. У хворих звичайно переважає не порушення інтелекту, а відхилення в емоційно-вольовій сфері по типу інфантилізму (дитячості). Їм властиві незрілість думок, нестійкість уваги, низька працездатність через значну стомлюваність, підвищена навіюваність, слабка ініціативність, нездатність до тривалої вольової дії. Хворі на синдром Клайнфельтера схильні до повчальних, часто пустих розмірковувань. У них карикатурна дорослість мови, майже постійна незадоволеність, схильність до бурчання.
Як і у разі синдрому Шерешевського-Тернера, протягом періоду статевого дозрівання виявляються невротичні реакції (найчастіше через усвідомлення своєї патології), що носять виражений та стійкий характер. Поведінка хворих на синдром Клайнфельтера характеризується поганим настроєм аж до депресивного стану, дратівливістю, нав’язливістю думок. Нерідко виявляються статеві збочення (гомосексуалізм).
6.2.2. Розумова відсталість, викликана хромосомними абераціями
Спадкові хвороби, спричинені порушеннями будови хромосом, теж характеризуються вадами розумового розвитку. Серед них найвідомішими є синдром Вільямса – Бейрена, синдром Лежена, синдром Вольфа – Хіршхорна та інші.
Найхарактернішими ознаками синдрому Вільямса – Бейрена (синдром «обличчя ельфа») є незвичайне обличчя, звуження надклапанної частини аорти або легеневої артерії, підвищений вміст кальцію в плазмі крові.
Популяційна частота захворювання варіює в межах 1:10000-25000 новонароджених незалежно від статі. Патологія спричинена делецією в довгому плечі 7-ої хромосоми (7q11.23), яка обіймає близько 15 генів.
Основними клінічними ознаками синдрому Вільямса – Бейрена є короткий ніс з відкритими вперед ніздрями, широка верхня і вузька нижня щелепа, надто широкий рот, повні щоки, відкопилені вуха, епікант, малий зріст. (Мал. 64).
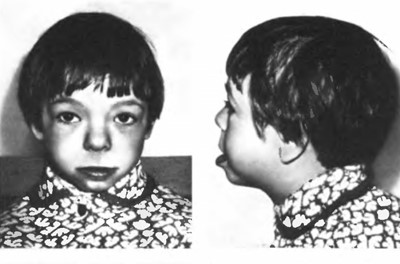
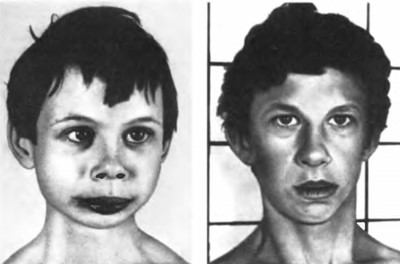
Мал. 64. Синдром Вільямса – Бейрена
Майже всі хворі відзначаються інтелектуальною недостатністю на рівні імбецильності (IQ в межах 30-50 балів). Вони звичайно досить балакучі, користуючись великим словарним запасом, але їх мова відзначається набором штампів, які часто вживаються невпопад, без зв’язку з ситуацією. Більшість із них має гарний музичний слух.
Діти з синдромом Вільямса – Бейрена, як правило, доброзичливі, привітні, слухняні. Спостерігаються різноманітні неврозоподібні розлади – страхи, нав’язливі думки, енурез тощо.
Специфічної терапії цього захворювання не існує, тому основне місце тут займає симптоматичне лікування та корекційно-виховна робота.
У шкільному навчанні хворі діти менш успішні, ніж можна сподіватися, виходячи з їх особистісних якостей та рівня інтелекту. Причинами тут є труднощі організації навчально-трудового процесу, підвищена стомлюваність та пересичливість хворих дітей.
Синдром Лежена та синдром Вольфа – Хіршхорна описані вище (див. розділ 5.5.1) і через тяжкість розумової відсталості та незначний термін життя хворих тут не розглядаються.
До спадкових патологій, спричинених хромосомними абераціями, можна віднести також ряд хвороб імпринтингу, серед яких найвідомішими є спадкові синдроми Прадера – Віллі та Ангельмана (див. розділ 5.6.1).
Синдром Прадера – Віллі. Майже всі хворі на синдром Прадера – Віллі розумово відсталі, але значення їх коефіцієнта інтелекту може варіювати в широкому діапазоні – від 20 до 90. Мова утруднена з незначним словарним запасом. Часто через підвищену пересичливість і стомлюваність хворих інтелектуальні труднощі здаються більш вираженими, ніж вони є насправді,. Якщо враховувати ці особливості в навчальному процесі, то можна досягти значно більших успіхів порівняно з очікуваними.
Хворі на синдром Прадера – Віллі, як правило, доброзичливі, але безініціативні, з частими різкими невмотивованими змінами настрою. У ряді випадків у хворих виявляються аутичні риси характеру, що може привести до помилкового діагнозу «аутизм».
Для дорослих хворих на фоні розумової відсталості різного ступеня тяжкості характерні емоційна нестійкість та низька пізнавальна здатність.
Хворі на синдром Ангельмана відзначаються тяжкою розумовою відсталістю аж до рівня ідіотії, невмотивованою веселістю, частим висуванням язика, грубою затримкою мовного розвитку.
6.2.3. Розумова відсталість при моногенних хворобах
До групи моногенних патологій з розумовою відсталістю відносяться деякі спадкові хвороби обміну речовин, хвороби сполучних тканин, окремі форми мікроцефалії, гідроцефалії та ряд інших захворювань.
Як було відзначено вище (див. розділ 5.4.2), серед моногенних захворювань численну групу складають спадкові дефекти обміну, і зокрема ензимопатії, або ферментопатії. На сьогодні відомо понад 100 ензимопатій, із яких майже для половини принципово розроблені методи медикаментозного чи дієтологічного лікування. Рання діагностика і своєчасно почате лікування в більшості випадків забезпечують попередження ураження мозку, а отже і розумову відсталість, на самих уразливих етапах його формування. Успадковуються ензимопатії за аутосомно-рецесивним або Х-зчепленим рецесивним типом. Їх популяційна частота варіює в широких межах – від 1:1000 до 1:1000000 новонароджених.
Визначальною ланкою ензимопатії є відсутність або значне зниження активності певного ферменту, що блокує або викликає істотне порушення того чи іншого біохімічного процесу. Оскільки більшість ферментних систем багатокомпонентні, ензимопатії звичайно представлені кількома генетичними формами. Слід також врахувати, що фермент практично завжди приймає участь у кількох метаболічних ланцюгах, внаслідок чого захворювання вражає кілька систем органів, тобто є полісимптомним. У таких випадках ізольовані порушення інтелекту зустрічаються рідко, але особливо часто вражається зір (при галактоземії, гомоцистинурії, мукополісахаридозах, амавротичній ідіотії тощо).
Однією із поширених і добре вивчених ензимопатій є фенілкетонурія (див. розділ 5.4.2.). Головним наслідком цього захворювання є розумова відсталість: в 65% – глибока, в 32% – значна та помірна і лише в 3 % – легка. Це корелює з такими морфологічними ознаками як вторинна мікроцефалія, мала маса мозку, дефекти мієлінізації в корі великих півкуль (особливо в лобових і скроневих зонах) та інших нервових структурах (наприклад, у зорових провідних шляхах).
У дітей, хворих на фенілкетонурію, виявляються значні порушення у розвитку мови: вони або зовсім нездатні говорити, або вживають окремі слова, які не пов’язані з об’єктами. Звуковідтворення та розуміння мови оточуючих порушене.
Відзначається погана координація рухів, часто трапляються епілептиформні напади.
Поведінка хворих різна. В одних випадках вона характеризується руховим неспокоєм, безцільними маніпуляціями з предметами і т.п. В інших – діти пасивні, мляві, позбавлені відчуття прихильності, погано впізнають близьких, пожвавлюються головним чином при згадці про їжу.
Порушення інтелектуального розвитку виявлено також у деяких гетерозигот – носіїв аномального гена. У 4 % випадків виявлена легка інтелектуальна недостатність, а в 6,5 % – нижня межа норми за інтелектом з відповідним невисоким рівнем освіти, професійної та соціальної адаптації.
Вчасна дієтотерапія дозволяє приблизно в 90 % випадків попередити розвиток розумової відсталості. Якщо лікування починається в старшому віці, розвиток інтелектуальної недостатності попередити не вдається, але поведінка дещо нормалізується, рідше трапляються епілептоподібні напади.
При деяких захворюваннях (їх називають «хворобами накопичення») пошкоджені ферменти розпаду деяких речовин, внаслідок чого останні накопичуються в клітинах, порушуючи всю їх життєдіяльність. Прикладом таких патологій можуть бути хвороба Німана – Піка, мукополісахаридози та інші.
У випадку хвороби Німана – Піка виявляється дефіцит ферменту розпаду ліпіду сфінгомієліну. Продукти неповного розпаду цього ліпіду накопичуються в клітинах печінки, селезінки, головного мозку, лімфатичних вузлах, лімфоцитах. Хвороба Німана – Піка спричинюється мутацією гена сфінгомієлінази (SMPD1), локалізованого в короткому плечі 11-ої хромосоми (11р15.4-15.1) і успадковується за аутосомно-рецесивним типом. Виділяють декілька форм захворювання, але всі вони характеризуються швидким наростанням тяжких неврологічних розладів і порушень фізичного та розумового розвитку. Хворі гинуть у віці 3-5 років. Середня розповсюдженість хвороби Німана – Піка складає 1:250000 новонароджених. Лікування симптоматичне.
До спадкових захворювань з розумовою відсталістю відносять і синдром Мартіна – Белла (див. розділ 5.6.2.). Це – Х-зчеплена рецесивна патологія, яка виявляється переважно у хлопчиків.
У всіх випадках синдрому Мартіна – Белла виявляється розумова відсталість, проте глибина її різна. Так, для хворих хлопчиків ступінь розумової аномалії може варіювати від помірного до глибокого (коефіцієнт інтелекту 70-35), в той же час у осіб жіночої статі з цією патологією виявляється лише легка інтелектуальна недостатність.
Мова хворих на синдром Мартіна – Белла часто поспішна, неритмічна, перекручена, з порушенням наголосів, нерозбірлива, з частими повторами та труднощами у підборі слів.
Трапляються окремі симптоми дитячого аутизму та шизофренії. Навіть у тих випадках, коли в клінічній картині провідною ознакою є інтелектуальні порушення, деякі з властивих аутизму особливостей (вразливість, значна здатність до пересичення, низька спроможність спілкування, іноді наявність особливих інтересів) вельми істотні, і їх необхідно враховувати в психологопедагогічній роботі.
Динаміка хвороби інколи виявляє тенденцію до зниження інтелектуального рівня, що, проте, вимагає спеціального вивчення. Адже, крім біологічних причин, на перебіг патології можуть суттєво впливати неадекватні методи навчання та виховання, вживані протягом кількох років. У зв’язку з цим суттєве значення у роботі з хворими на синдром Мартіна-Белла має поетапна педагогічна корекція.
Серед різноманітних форм розумової відсталості моногенної природи виділяють так звані ксеродермні форми, коли інтелектуальний дефект поєднується з ураженням шкіри. Прикладом таких патологій може бути нейрофіброматоз та туберозний склероз.
Для нейрофіброматозу характерна наявність множинних пухлин центральної та периферійної нервової системи, органів зору, внутрішніх органів, аномалії кісток та пігментації шкіри, родимі плями.
Виразна форма нейрофіброматозу зустрічається з частотою 1:25001:3000 новонароджених, успадковується за аутосомно-домінантним типом. даний час виділяють дві форми нейрофіброматозу: класичну периферійну (нейрофіброматоз I, або хвороба Реклінгаузена), ген якої (NF1) локалізований у 17-й хромосомі (17q11.2), та центральну форму (нейрофіброматоз II), ген якої (NF2) знаходиться в 22-й хромосомі (22q12.2).
Захворювання на нейрофіброматоз І виявляється з моменту народження або в перше десятиріччя життя. Визначальною та найбільш ранньою діагностичною ознакою цієї хвороби є висипання дрібних плям кольору кави в пахвових западинах. Для хворих характерні вади будови обличчя та тіла: голова, як правило, велика та деформована, риси обличчя грубі, збільшена відстань між парними органами, очні щілини та вушні раковини деформовані, кисті рук та стопи великі та широкі, шия коротка, грудна клітка дуже спотворена.
Ураження нервової системи різноманітні за спектром, проявом і динамікою, що визначається локалізацією та розміром новоутворень. Проявами їх є зниження інтелекту, порушення пам’яті, уваги, іноді судоми. Ці ознаки можуть виявлятися не у всіх хворих. Починаються вони з незначних симптомів, але, поступово наростаючи, приводять до розладів мови, ослабленню окремих вищих психічних функцій і, як наслідок, до труднощів навчання. З часом у багатьох випадках шкільні проблеми наростають, ускладнюються індивідуальними аномаліями і можуть привести до переводу на більш низький або індивідуальний рівень навчання.
Характерною особливістю нейрофіброматозу II типу є утворення пухлин черепно-мозкових нервів і спинного мозку. В клінічній картині, перш за все, виявляються різні неврологічні розлади, прогресуюче зниження інтелекту та повний розпад психіки. При цьому, як правило, пухлини на шкірі та периферійних нервах не розвиваються. Для корекційної педагогіки цей тип захворювання істотного значення не має.
Лікування нейрофіброматозу симптоматичне. Наявні пухлини видаляються хірургічним шляхом.
6.2.4. Мультіфакторіально обумовлена розумова відсталість
Ця форма розумової відсталості вважається досить поширеною, однак вивчена недостатньо. Вважають, що вона проявляється внаслідок адитивної (сумарної) дії багатьох генів (спадкова складова, або схильність) і чинників середовища (неспадкова складова). Така розумова відсталість, як правило, не супроводжуються неврологічними розладами та виразними морфологічними відхиленнями. При цьому інтелектуальна недостатність практично завжди легка та неускладнена за структурою.
Таким чином, спадково обумовлена розумова відсталість різноманітна за клінічними проявами та генетичною природою. При цьому зовнішні чинники втричі рідше, ніж генетичні, є безпосередньою причиною порушень інтелектуального розвитку, однак вони можуть спричинити прояв генетичної патології.
6.2.5. Реабілітація хворих із розумовою відсталістю
Проблеми лікування та реабілітації розумово відсталих дітей тісно взаємопов’язані та охоплюють широке коло медичних, педагогічних і соціальних заходів. Організація всебічної допомоги таким дітям повинна здійснюватися установами охорони здоров’я, соціального забезпечення, загальної та професійної освіти.
Найважливішою умовою реабілітації розумова відсталих дітей є раннє виявлення недуги, своєчасне і поетапне надання лікувальної та корегувальнопедагогічної допомоги, яку має здійснювати мережа відповідних установ, диференційованих з урахуванням віку та ступеня розумових вад. Ця мережа включає спеціалізовані дошкільні заклади, допоміжні школи та школиінтернати для виховання та навчання олігофренів шкільного віку, спеціалізовані професійно-технічні училища для розумово відсталих підлітків, а також інтернати соціального забезпечення для хворих з глибокою розумовою відсталістю.
Лікувально-педагогічна робота повинна формуватися для кожного конкретного випадку з урахуванням клінічної картини захворювання, структури інтелектуального дефекту, темпераменту, мови та моторики хворих. Велике значення в поліпшенні нервово-психічного розвитку дітей-олігофренів мають логопедичні заходи, спрямовані на формування мовних функцій та усунення дефектів мови. При інтелектуальній недостатності, що супроводжується соматичними, неврологічними та руховими аномаліями, велике значення має лікувальна фізкультура, сприяюча розвитку моторики, координації рухів, уваги, емоційної сфери, зміцненню загального фізичного стану хворих.
Навчально-виховна робота в допоміжній школі-інтернаті включає початкові етапи трудового навчання, яке займає одне з основних місць в процесі підготовки розумово відсталих дітей до самостійної суспільно-корисної діяльності. Соціально-трудова адаптація осіб з вадами розумового розвитку має ряд специфічних особливостей, вимагає спеціальних послідовних прийомів і методів професійного навчання з подальшим поступовим включенням підлітків у самостійну трудову діяльність.