Генетика людини: методичка
5.3. Загальна характеристика спадкових патологій
Не дивлячись на різноманіття клінічної картини спадкових захворювань, можна виділити їх загальні риси, які дадуть підстави стверджувати або заперечувати наявність патології у обстежуваної людини. Основою формування загальних клінічних характеристик різних форм спадкових патологій є генетичний контроль ключових ланок обміну речовин та морфогенних процесів.
Первинною ланкою перебігу спадкових патологій є генні мутації та порушення структури або кількості хромосом.
Особливості патогенезу спадкових захворювань в основному визначаються трьома чинниками: характером пошкодження спадкових структур, генетично визначеними морфо-фізіологічними особливостями організму та дією факторів навколишнього середовища. Ці чинники і обумовлюють індивідуальний характер перебігу патологічних процесів у кожному конкретному організмі.
Хоча спадкові захворювання, як і будь-які інші хвороби, мають свої характерні риси, жодну з приведених нижче особливостей не можна розглядати як щось абсолютне, бо тільки наявність характерних рис в сукупності дозволяє прогнозувати спадкову патологію у пацієнта.
5.3.1. Основні властивості спадкових патологій
Генні патології. Мутаційна зміна нуклеотидної послідовності у молекулі ДНК є причиною моногенних спадкових хвороб. Специфіка патогенезу моногенних захворювань визначається особливостями хімічної природи первинного продукту гена, обумовленими конкретною мутацією, і тією роллю, яку цей продукт грає в життєдіяльності організму. При одних мутаціях, що обумовлюють повну відсутність необхідної організму речовини (наприклад, соматотропного гормону або цитохрому-450), нормальний розвиток організму або утруднений, або неможливий. За інших мутацій, що приводять до дефіциту біологічно активної речовини або структурного білка, виникають захворювання, що характеризуються розладом структури та функції окремих тканин, органів або фізіологічних систем.
Описані варіанти патогенезу моногенних (менделюючих) захворювань надзвичайно різноманітні, що переважно визначається величезним числом порушень біохімічних реакцій, які здійснюються в організмі. Не дивлячись на це, виділені деякі загальні закономірності розвитку моногенних форм патології. Наприклад, для багатьох спадкових хвороб обміну речовин визначено прямий зв’язок між мутантним геном і порушеною біохімічною реакцією.
Дотепер відкриті та детально описані сотні видів спадкових аномалій метаболізму, спричинених мутацією одного гена. Обумовлена мутацією гена аномалія амінокислотної послідовності поліпептидного ланцюга істотно порушує активність ферментів. Переважна більшість випадків спадкових патологій обміну пов’язані саме із зміною активності ферментів, що призводить до розладу або зупинки реакцій у даному ланцюзі метаболізму, викликаючи розвиток тієї або іншої хвороби. При цьому захворювання може виникати внаслідок таких причин: 1) накопичення субстрату, або надлишку речовини (наприклад, цереброзиду при хворобі Гоше), яка підлягає дії ферменту; 2) збільшення вмісту речовини-попередника (наприклад, метіоніну при цистатионінурії); 3) недостатнє утворення речовини (зокрема, дефіцит цитидінтрифосфату при оротовій ацидурії); 4) збільшення концентрації токсичних продуктів метаболізму (наприклад, фенілацетилглютаміну, фенілоцтової, фенілпіровиноградної кислоти та інших фенілкетонових похідних при фенілкетонурії).
Проте навіть у випадках схожості виду порушень, наприклад при накопиченні субстрату, механізми розвитку різних захворювань будуть різні. В одному випадку субстрат (наприклад, високомолекулярна речовина у разі амавротичної ідіотії Тея – Сакса), що нагромаджується, може відкладатися в клітинах, поступово приводячи їх до загибелі. В інших випадках надлишок низькомолекулярної розчинної речовини створює високу її концентрацію в біологічних рідинах організму. Наприклад, при галактоземії (внаслідок недостатності галактозо-1-фосфат-уридилтрансферазы) відбувається накопичення глюкозо-1-фосфата, токсичні концентрації якого ушкоджують різні тканини, у зв’язку з чим виникають цироз печінки, катаракта, ушкоджуються нирки, нейрони головного мозку.
Хромосомні патології. Характерною особливістю патогенезу хромосомних хвороб є раннє порушення морфогенезу. Воно виявляється розладом поділу та дозрівання клітин, порушенням їх міграції та диференціації, що обумовлює виникнення множинних аномалій формування різних органів і тканин. В основі спостережуваних явищ лежить, як вважають, незбалансованість геному.
Розрізняють три типи ефектів у процесі розвитку хромосомних хвороб: специфічні, напівспецифічні та неспецифічні. Специфічні ефекти залежать від зміни кількості структурних генів (унікальних для будь-якої хромосоми), що кодують синтез того чи іншого білка. При трисоміях число їх збільшується, а при моносоміях – зменшується. Напівспецифічні ефекти при хромосомних хворобах обумовлені зміною числа генів, представлених в геномі великою кількістю копій (наприклад, гени рибосомних, гістонових, скоротливих білків і т.п.). Неспецифічні прояви при хромосомних абераціях можуть бути обумовлені зміною структури гетерохроматину (генетично інертних ділянок хромосом), який виконує важливу роль у процесах клітинного поділу, росту тощо.
У випадку хромосомних аберацій прояв відхилення від нормального розвитку, як правило, корелює із ступенем хромосомного дисбалансу. Чим більше хромосомного матеріалу залучено до аберації, тим раніше захворювання виявиться в онтогенезі і тим значнішим буде порушення фізичного та психічного розвитку людини. При цьому надлишок хромосомного матеріалу менше впливає на клінічну картину хвороби порівняно з його втратою. Наприклад, втрата однієї з аутосом перешкоджає імплантації яйцеклітини в стінку матки. В той же час відомі значні хромосомні синдроми, обумовлені трисоміями різних хромосом.
Надлишок або нестача гетерохроматинових ділянок хромосом можуть і не мати клінічних наслідків. На відміну від цього втрата еухроматинових (генетично активних) ділянок хромосом завжди приводить до втрати унікальних генів та до розвитку тяжких патологій.
5.3.2. Вік прояву спадкових захворювань
Більшість спадкових хвороб розпізнається в перинатальному (під час та одразу після пологів) та ранньому дитячому віці. Як правило, перші симптоми захворювання діагностуються з моменту народження або невдовзі після нього. Наприклад, деякі спадкові синдроми, що супроводжуються вадами розвитку (розщеплення губи або піднебіння, додаткові пальці на кисті та стопі, відсутність кінцівки, дефекти передньої черевної стінки, пупкова грижа тощо), можуть бути виявлені при народженні дитини. Прикладами раннього прояву (або вродженого характеру) спадкових хвороб є всі хромосомні синдроми, ахондроплазія та ряд інших аномалій скелета. Проте в деяких випадках перші клінічні прояви спадкового захворювання можуть виявлятися і в більш старшому віці.
Спостереження показали, що 25 % усіх моногенних хвороб розвиваються до народження. Приблизно до 3 років виявляється ще 50 % таких захворювань. Разом з тим відомі спадкові захворювання з пізніми термінами прояву (хвороба Альцгеймера, хорея Гентинґтона та інші).
Необхідно також підкреслити, що є цілий ряд відмінностей у патологій, які проявляються в різні вікові періоди життя людини (табл. 8). Наприклад, з віком зменшується частота моногенних хвороб і збільшується частка мультифакторних (багаточинникових), прояв яких провокується чинниками навколишнього середовища. Це можна пояснити тим, що з віком знижується роль спадкових факторів у розвитку патологій.
Таблиця 8. Деякі властивості спадкових патологій залежно від віку прояву
Віковий період | До настання статевої зрілості | Після настання статевої зрілості |
Тип успадкування | Моногенний | Мультифакторний |
Вік початку | Ранній | Пізній |
Частота | Рідкісні | Часті |
Період до прояву хвороби | Короткий | Довгий |
Кількість хворих родичів | Багато | Набагато менше |
Кількість хвороб | Дуже велика | Значно менша |
Відмінності, зумовлені статтю | Рідкісні | Часті |
Успадковуваність | Висока | Низька |
Прогнозованість | Висока | Низька |
5.3.3. Прогредієнтність та хронічність спадкових патологій
Прогредієнтним називається перебіг захворювання з постійним погіршенням загального стану та з наростанням негативних симптомів у пацієнта. Для багатьох спадкових хвороб характерне поступове наростання їх тяжкості по мірі розвитку патологічного процесу. Наприклад, при фенілкетонурії у процесі росту дитини з’являються і посилюються симптоми затримки психомоторного та розумового розвитку, формується вторинна мікроцефалія. Хвороба Тея – Сакса починається з 6-місячного віку демієлінізацією нервових волокон, що поступово приводить до летального наслідку.
Спадкові хвороби, що починаються в будь-якому віці, мають хронічний характер. Наприклад, діти, хворі на спінальну аміотрофію Вердніґа – Гофмана, поступово втрачають рухову активність у результаті загибелі мотонейронів передніх рогів спинного мозку, а у хворих з легеневою формою муковісцидозу формується хронічна пневмонія через підвищену в’язкість секретів бронхіальних слизових залоз. У випадку гомозиготної β-таласемії на фоні наростаючого недокрів’я поступово збільшуються розміри печінки та селезінки, розвиваються аномалії кісткової та імунної системи. Численні форми спадкових патологій були виявлені саме при обстеженні пацієнтів з хронічним перебігом захворювання.
Генетичною основою прогредієнтності та хронічного перебігу спадкових хвороб є, як правило, безперервність дії патологічного гена та відсутність його нормального продукту, що приводить до порушення нормального функціонування клітин, тканин і органів.
Ступінь цих явищ для одного і того ж захворювання у різних пацієнтів може розрізнятися.
5.3.4. Ураження багатьох органів і систем при спадкових захворюваннях
Практично для всіх форм спадкової патології характерна множинність ураження. В першу чергу це обумовлено плейотропною дією гена, тобто його здатністю контролювати розвиток різних ознак організму. Наприклад, при синдромі Марфана уражуються кісткова, серцево-судинна системи та органи зору; при синдромі Лоренса – Муна уражені кісткова, сечостатева, ендокринна системи та органи зору; при галактоземії патологією охоплені печінка, ЦНС, зір.
Механізми первинної плейотропної дії генів при деяких спадкових захворюваннях уже відомі. Це, як правило, хвороби обміну речовин, у тому числі хвороби накопичення. Так, при аутосомно-рецесивному захворюванні Вестфаля – Вільсона – Коновалова дефект сироваткового білка церулоплазміну не забезпечує ефективний транспорт міді. Це спричинює відкладення її надлишку в різних органах і тканинах, що викликає множинні ураження.
5.3.5. Сімейний характер спадкових патологій
Більшість спадкових захворювань характеризується наявністю їх у членів однієї сім’ї. На відміну від інфекційних захворювань розподіл випадків спадкових патологій у поколіннях та залежно від статі підлягає закономірностям Г. Менделя.
Разом з тим наявність захворювання тільки у одного з членів родоводу не виключає спадкового характеру цієї хвороби. Подібна ситуація може бути обумовлена такими причинами: а) наявністю нової домінантної мутації, що відбулася в аутосомі чи в статевій хромосомі одного з батьків, б) явищем неповної пенетрантності домінантного гена, в) гетерозиготністю обох батьків за патологічним рецесивним геном, г) наявністю рецесивної Х-зчепленої патології.
5.3.6. Специфічні симптоми спадкових захворювань
Наявність рідкісних специфічних симптомів або їх поєднань у хворого дає підстави думати про спадкову або природжену природу його захворювання. Наприклад, у дитини з розщепленням піднебіння наявність симетричних заглибин або свищів на слизовій оболонці нижньої губи дозволяє підозрювати аутосомно-домінантний синдром Ван дер Вуда; наявність широкого 1-го пальця кистей та стоп у дитини з прогресуючою розумовою відсталістю наводить на думку про аутосомно-домінантний синдром Рубінштейна – Тейбі тощо.
У деяких випадках симптоми прояву генів, що не мають ніякого клінічного значення, є опорними при визначенні патології. Наприклад, виявлення насічок на мочці вуха у дитини з надмірно великим язиком та розходженням прямих м’язів живота є вирішальним для визначення синдрому Беквіта – Відемана і, отже, виправданого вибору відповідної терапії.
5.3.7. Резистентність спадкових хвороб до лікування
Говорячи про резистентність (стійкість) спадкових хвороб до лікування, потрібно пам‘ятати про відносність цього поняття.
По-перше, ця клінічна особливість пояснюється тим, що у багатьох випадках виправити первинні ланки патогенезу захворювань, навіть якщо відомий первинний продукт мутантного гена, не завжди вдається. По-друге, явище резистентності часто спричинюється тим, що симптоми спадкових захворювань сприймаються як саме захворювання.
Цим пояснюється безуспішність використовування стандартних схем, підходів і методів терапії. Наприклад, лікування екземи, яка є шкірним проявом протокопропорфірії (спадкова хвороба крові), часто полягає у лікуванні симптому, тоді як для даного захворювання розроблені надійні методи профілактики та терапії, засновані на закономірностях його патогенезу. При нерозпізнаному синдромі Костмана інтенсивна антибіотикотерапія гнійничкових виразок слизистих оболонок і шкіри не рятує дитини від прогресуючого перебігу хвороби, що виявляється у вигляді фурункулів, нагноєнь підшкірної клітковини, важких стоматитів, блефаритів (запалення країв повік) і т.п.
Важливо підкреслити, що будь-яка окремо взята з відомих клінічних особливостей не може бути абсолютним критерієм для визначення спадкового захворювання. Проте сукупність декількох характерних для даної спадкової патології особливостей може слугувати достатньою основою для її визначення.
5.3.8. Поліморфізм спадкових патологій
Під поліморфізмом розуміється численність клінічних ознак та лабораторних показників будь-якого захворювання, а також різноманітність їх прояву. Наприклад, у частини хворих на синдром Марфана можна діагностувати випинання мітрального клапана серця, а у інших – аневризму (розслаблення та випинання стінки) аорти. З боку органів зору може виявлятися підвивих кришталика, а може мати місце незначна короткозорість тощо.
Іншим прикладом різноманітного прояву симптоматики при одному й тому ж захворюванні є нейрофіброматоз I типу (наявність численних пігментних плям, шкірних та підшкірних пухлин). Відомі випадки, коли у одного хворого на нейрофіброматоз спостерігається повна клінічна картина, для якої характерні множинні пухлини, тоді як у іншого хворого (навіть у члена тієї ж сім’ї) реєструються лише пігментні плями.
Описані вище явища спричинюються такими особливостями генів, у тому числі й мутантних, як пенетрантність та експресивність (див. розділ 4.1.1.). Згадаємо, що пенетрантність – це вірогідність, або частота прояву захворювання (ознаки) у носіїв даного гена. Експресивність же визначається як ступінь прояву дії певного гена.
Якщо та чи інша ознака визначається лише наявністю відповідного гена, то говорять про 100%-ну пенетрантність цього гена. Коли ж за наявності гена захворювання виявляється не в усіх випадках, то говорять про неповну пенетрантність.
Одне і те ж захворювання може по-різному протікати у різних хворих, навіть у членів одного родоводу. Тоді говорять про різну експресивність гена, яка і має місце у другому з описаних вище випадків. Іноді прояв гена буває настільки незначним, що ознаки його не виявляються існуючими нині діагностичними методами.
Клінічний поліморфізм спадкових хвороб, визначуваних одним геном, може проявитися різним часом початку захворювання або різною тяжкістю клінічних проявів. Наприклад, хорея Гентинґтона, середній вік початку якої приблизно 40 років, може початися в дитячому віці, а в деяких випадках – лише після 60 років.
5.3.9. Генетична гетерогенність патологій
Коли клінічно схожі хвороби в різних сім’ях обумовлені дефектами різних генів, говорять про генетичну гетерогенність спадкових патологій. Вживані назви багатьох патологічних станів або навіть захворювань часто приховують властиву їм гетерогенність, надаючи вигляду однорідності. За кожною, наприклад, з таких назв, як «синдроми кровоточивості», «м’язова дистрофія», «розумова відсталість», «глухота», «анемія», «глікогеноз» (аномальне накопичення глікогену) і т.д., ховається кілька клінічно, біохімічно та генетично різних захворювань. Фенотипно (клінічно) однорідне захворювання може бути генетично гетерогенним, якщо його ланцюг біохімічних реакцій блокований в різних точках, як, наприклад, при адреногенітальному синдромі (порушення біосинтезу гормонів). Відсутність або нестача одного й того ж кінцевого продукту, наприклад гемоглобіну, що приводить до недокрів’я, також може бути наслідком мутацій різних генів.
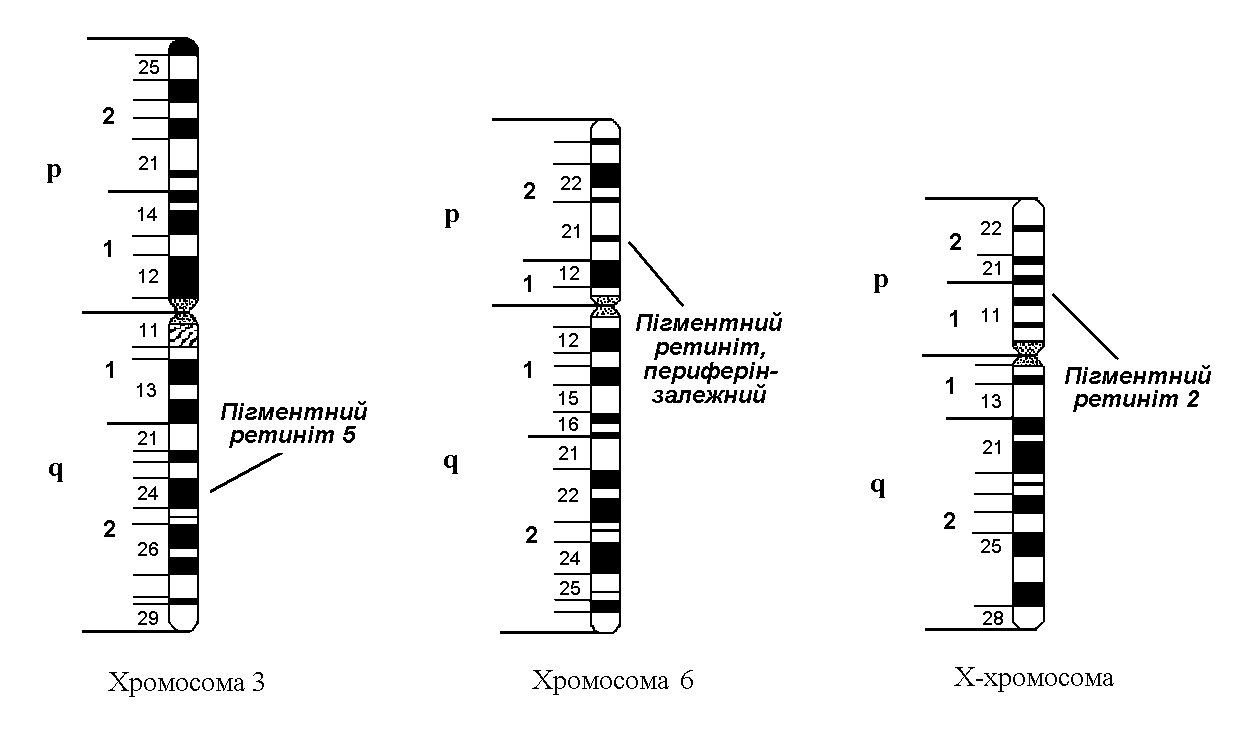
Мал. 37. Локалізація генів деяких форм пігментного ретиніту на хромосомах
Нижче подаються ознаки гетерогенності спадкового захворювання.
- Наявність сімей з різними типами успадкування клінічно схожих захворювань. Наприклад, пігментний ретиніт (зниження нічного зору, звуження полів зору з подальшою прогресуючою втратою зору до повної сліпоти) може успадковуватися як аутосомно-домінантний, або як аутосомнорецесивний, або як Х-зчеплена рецесивна ознака. Показано, що ген аутосомно-домінантної форми локалізований на довгому плечі хромосоми 3 (3q24), ген аутосомно-рецесивної форми – на короткому плечі хромосоми 6 (6p21.3), а ген Х-зчепленої форми – на короткому плечі Х-хромосоми (Xp11.3) (мал. 37). Цікаво, що, крім щойно названих форм пігментного ретиніту, виявлено ще принаймні 9, аномальні гени яких локалізовані в хромосомах 1, 7 (2 гени), 8, 17, 19 та Х (3 гени, крім названого).
- Народження дітей (див. розділ 4.1.2, мал. 32, IV покоління) з нормальним слухом від батьків, які обоє хворі на аутосомно-рецесивну глухоту різних форм. Сьогодні виявлено значну кількість форм глухоти з аутосомнорецесивним типом успадкування, спричинених мутаціями у генах, локалізованих, наприклад, у хромосомах 2, 3, 4, 7, 13, 14, 17, 21 та інших.
- Наявність різних провідних біохімічних дефектів. Так, при синдромі Санфіліппо (порушення вуглеводного обміну, що супроводжується важкими фізичними вадами та розумовою відсталістю) виділяють 4 типи біохімічних дефектів. Кожний з цих типів характеризується браком того чи іншого ферменту, який бере участь у вуглеводному обміні на певній його стадії.