Лімфопроліферативні захворювання
Пухлини лімфатичної системи включають гострий лімфобластний лейкоз (вивчають в курсі гематології), хронічний лімфолейкоз, неходжкінські лімфоми, лімфому Ходжкіна та парапротеїнемічні гемобластози (мієломна хвороба, макроглобулінемія Вальденстрема, хвороба важких ланцюгів).
КОРОТКИЙ АНАТОМО-ФІЗІОЛОГІЧНИЙ НАРИС
Лімфатична система (system lymphaticum) — система лімфатичних капілярів, дрібних і великих лімфатичних судин, а також лімфатичних вузлів, що містяться вздовж цих судин. Ця система разом із венами забезпечує дренажну функцію, тобто всмоктування із тканин води, колоїдних розчинів білків, емульсій ліпідів, розчинених у воді кристалоїдів, видалення із тканин продуктів розпаду клітин, мікробних тіл та інших частинок, а також лімфоцитопоетичну і захисну функції.
На відміну від кровоносної системи, лімфатична не є закритою і не має центрального насоса. Лімфа, що циркулює в ній, рухається повільно і під невеликим тиском.
У структуру лімфатичної системи входять:
- лімфатичні капіляри;
- лімфатичні судини;
- лімфатичні вузли;
- лімфатичні стовбури і протоки;
- селезінка.
Лімфатичні вузли (nodi lymphatici) — периферичні органи імунної системи, що виконують функцію біологічних фільтрів, а також функції лімфоцитопоезу і утворення антитіл. Будову лімфатичного вузла наведено на рис.18.1.
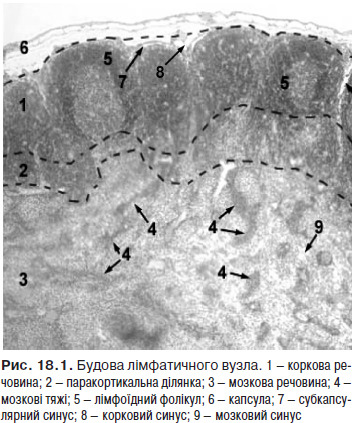
На рис. 18.2 зображено групи лімфатичних вузлів.

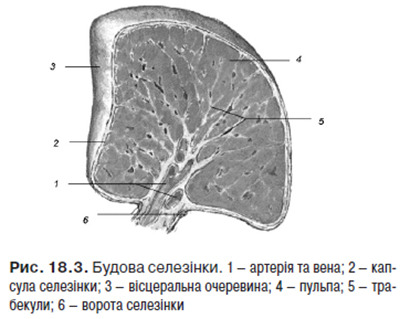
Селезінка (lien, spleеn) — непарний, видовженої форми периферичний орган лімфоїдного кровотворення та імунного захисту. Основні функції селезінки: елімінація еритроцитів і тромбоцитів, які завершили свій життєвий цикл, депонування крові та заліза, розмноження і антигензалежна диференціація лімфоцитів та утворення антитіл, вироблення біологічно активних речовин, які пригнічують еритропоез у червоному кістковому мозку, в ембріональний період селезінка є універсальним кровотворним органом, в якому утворюються всі формені елементи крові. Будову селезінки зображено на рис. 18.3.
Лімфопроліферативні захворювання — гетерогенна група злоякісних захворювань лімфатичної системи. Субстратом є В- та Т-лімфоцити, які знаходяться на різних етапах диференціації. Клінічно захворювання супроводжуються збільшенням лімфатичних вузлів та інших лімфоїдних органів, ураженням кісткового мозку. Захворювання суттєво відрізняються між собою за біологічними властивостями, клінічними проявами, морфологічними особливостями, відповіддю на терапію та прогнозом.
Загальноприйнятою на сьогодні є класифікація лімфоїдних новоутворень ВООЗ (2008), в основу якої покладено результати аналізу морфологічних, імунофенотипових, генетичних ознак та клінічних даних.
Класифікація лімфоїдних новоутворень ВООЗ (2008)
Пухлини зі зрілих (периферичних) В-клітин:
- Хронічний лімфолейкоз/лімфома з малих лімфоцитів
- В-клітинний пролімфоцитарний лейкоз
- В-клітинна лімфома маргінальної зони селезінки
- Волосатоклітинний лейкоз
- Лімфома з ураженням селезінки, некласифікована
- дифузна дрібноклітинна В-клітинна лімфома червоної пульпи селезінки
- варінт волосатоклітинного лейкозу
- Лімфоплазмоцитарна лімфома
- Макроглобулінемія Вальденстрема
- Хвороби важких ланцюгів
- хвороба α-важких ланцюгів
- хвороба γ-важких ланцюгів
- хвороба μ-важких ланцюгів
- Плазмоклітинна мієлома
- Солітарна кісткова плазмоцитома
- Позакісткова плазмоцитома
- Екстранодальна В-клітинна лімфома маргінальної зони MALT-типу
- Нодальна В-клітинна лімфома маргінальної зони
- Нодальна В-клітинна лімфома маргінальної зони дитячого віку
- Фолікулярна лімфома
- Фолікулярна лімфома дитячого віку
- Первинна лімфома із ураженням фолікулярних центрів шкіри
- Мантійноклітинна лімфома
- Дифузна В-великоклітинна лімфома
- В-великоклітинна лімфома, багата Т-клітинами/гістіоцитами
- первинна В-великоклітинна лімфома ЦНС
- первинна шкірна В-великоклітинна лімфома з локалізацією на нижніх кінцівках (leg type)
- EBV + дифузна В-великоклітинна лімфома людей похилого віку
- Дифузна В-великоклітинна лімфома асоційована з хронічним запаленням
- Лімфоїдний гранулематоз
- В-великоклітинна лімфома середостіння
- Інтраваскулярна В-великоклітинна лімфома
- ALK + В-великоклітинна лімфома
- Плазмобластна лімфома
- В-великоклітинна лімфома, що виникає при ННV8-асоційованій мультицентричній хворобі Кастлмана
- Первинна лімфома ексудатів
- Лімфома Беркіта
- Некласифікована В-клітинна лімфома з ознаками, проміжними між дифузною В-великоклітинною лімфомою та лімфомою Беркіта
- Некласифікована В-клітинна лімфома з ознаками, проміжними між дифузною В-великоклітинною лімфомою та класичною лімфомою Ходжкіна
Пухлини з Т-клітин та NК-клітин:
- Т-клітинний пролімфоцитарний лейкоз
- Т-великоклітинний лімфолейкоз із грануловмістких клітин
- Агресивний NК-лейкоз
- Системне ЕВV+ Т-лімфопроліферативне захворювання дитячого віку
- Гідроа оспеновидноподібна лімфома
- Т-клітинна лімфома/лейкоз дорослих (HTLV1+)
- Екстранодальна NК-/Т-клітинна лімфома, назальний тип
- Т-клітинна лімфома, асоційована з ентеропатією
- Гепатоспленічна Т-клітинна лімфома
- Підшкірна панікулоподібна Т-клітинна лімфома
- Грибоподібний мікоз
- Синдром Сезарі
- Первинне шкірне CD30+ Т-клітинне лімфопроліферативне захворювання
- Лімфоматоїдний папульоз
- Первинна шкірна анапластична великоклітинна лімфома
- Первинна шкірна γ/δ Т-клітинна лімфома
- Первинна шкірна CD8+ агресивна епідермотропна цитотоксична Т-клітинна лімфома
- Первинна шкірна CD4+ дрібноклітинна/середньоклітинна Т-клітинна лімфома
- Периферична Т-клітинна лімфома, некласифікована
- Ангіоімунобластна Т-клітинна лімфома
- Анапластична АLK+ великоклітинна лімфома
- Анапластична АLK- великоклітинна лімфома
Лімфома Ходжкіна:
- Нодулярна лімфома з переважанням лімфоцитів
- Класична лімфома Ходжкіна
- Нодулярний склероз
- Багата лімфоцитами
- Змішано-клітинний тип
- Лімфоїдного виснаження
ЛІМФОМА ХОДЖКІНА
Лімфома Ходжкіна — пухлинне захворювання, первинно локалізоване в лімфоїдній тканині, субстратом якого є гігантські багатоядерні клітини Березовського — Штернберга.
У 1832 р. Томас Ходжкін вперше описав 7 хворих, у яких була генералізована лімфаденопатія та ураження селезінки. У 1865 р. S. Wilks запропонував назву «хвороба Ходжкіна». У 1890 . С.Я. Березовський та в 1898 р. К. Штернберг описали великі багатоядерні клітини, характерні для уражених лімфатичних вузлів. У 1902 р. Д. Рід дала морфологічну характеристику клітин, що є субстратом хвороби. Згодом ці клітини отримали назву клітин Рід — Штернберга (в Росії — Березовського — Штернберга). Оскільки на сьогодні доведена лімфоїдна природа клітин Березовського — Штернберга з 2001 р. згідно з класифікацією ВООЗ використовують термін «лімфома Ходжкіна».
Лімфома Ходжкіна становить ≈1% усіх ЗН у мешканців розвинутих країн. В Україні у 2012 р. за даними Національного канцер-реєстру захворюваність на лімфому Ходжкіна становила 2,5 на 100 тис. дорослого населення, смертність — 0,8 на 100 тис. дорослого населення та виявлено 1126 первинних хворих на лімфому Ходжкіна. Чоловіки хворіють дещо частіше, ніж жінки. Захворюваність має два вікових піки. Перший пік характерний для осіб віком 15–40 років (максимум — 20–25 років), другий пік — віком старше 50 років.
ЕТІОЛОГІЯ
Згідно з сучасними уявленнями у розвитку лімфоми Ходжкіна провідну роль відіграють лімфотропні віруси. Так, відповідно до однієї з гіпотез, утворення клітин Березовського — Штернберга є результатом індукованого вірусом Епштейна — Барр злиття пухлинних Т-лімфоцитів або активування В-лімфоцитів. Дані фенотипування клітин лімфоми Ходжкіна підтверджують їх В-клітинне походження. Більшість сучасних дослідників дотримуються теорії про первинно вогнищеве виникнення лімфоми Ходжкіна. Початково вогнище ураження локалізується у тимусозалежній паракортикальній ділянці одного, іноді — декількох лімфовузлів. Пізніше процес розповсюджується на інші групи лімфовузлів по обидва боки діафрагми, на печінку, селезінку, кістковий мозок, кістки та ін. Прогресування процесу може супроводжуватися зміною гістологічних варіантів захворювання.
Основними факторами ризику розвитку лімфоми Ходжкіна є:
- Вік;
- Природжені порушення імунної системи;
- Ожиріння;
- М’ясо та жирна їжа;
- Радіація;
- Бензол, гербіциди та інсектициди;
- Набутий імунодефіцит, викликаний застосуванням різних препаратів у хворих із приводу пересадки органів;
- Інфекції, спричинені ВІЛ;
- Вірус Епштейна — Барр;
- Тютюнопаління.
КЛІНІЧНА КАРТИНА
Головним проявом лімфоми Ходжкіна є безсимптомне, однак помітне збільшення лімфатичних вузлів (лімфаденопатія). Наступними за частотою скаргами після лімфаденопатії є наявність так званих системних інтоксикаційних симптомів (В-симптомів), до яких належать прогресуюча втрата маси тіла на 10% за останні 6 міс, профузне пітніння, підвищення температури тіла >38 °С без наявності інфекційного захворювання чи інших причин, частіше у другій половині дня, яке може спонтанно зникати чи мати періодичний характер. У більшості випадків для лімфоми Ходжкіна характерним є генералізований свербіж шкіри різної інтенсивності, який раніше відносили до симптомів інтоксикації, внаслідок чого можна відмічати численні сліди подряпин. На сьогодні шкірний свербіж виключено з симптомів інтоксикації.
У хворих на лімфому Ходжкіна при огляді лімфатичні вузли здебільшого є твердими, малорухливими та неболючими, можуть утворювати конгломерати за рахунок поширення на сусідні локуси лімфатичної тканини, що є особливістю розповсюдження цієї пухлини в організмі. Своєрідний симптом лімфоми Ходжкіна — поява болю у лімфатичних вузлах при вживанні алкоголю. Серед периферичних груп лімфатичних вузлів найчастіше первинно уражуються нижньошийні та надключичні, дещо рідше — аксилярні, а також лімфатичні вузли в середніх та верхніх ділянках шиї. Нерідкою знахідкою при лімфомі Ходжкіна є ізольоване інтраторакальне збільшення лімфатичних мас (у 15% випадків), переважно переднього середостіння. Останні, як і конгломерати периферичних вузлів, нерідко досягають розмірів масивного пухлинного ураження (bulky disease), переважаючи своїм діаметром ⅓ поперечного розміру грудної клітки. У такому разі хворі часто скаржаться на сухий кашель, задишку. Може виникати симптом здавлення верхньої порожнистої вени. Якщо лімфоїдні конгломерати при масивному ураженні середостіння проростають плевру, перикард, трахею, легеневу тканину, то виникає перикардит і плеврит. При збільшенні лімфатичних вузлів черевної порожнини у хворого може виникати біль у животі, у попереку. У 25% випадків для лімфоми Ходжкіна характерне ураження селезінки переважно вогнищевого характеру, частіше без спленомегалії.
Серед органних уражень частіше відзначають залучення у патологічний процес легень. Ураження легеневої тканини має як вогнищевий, так і інфільтративний характер, іноді з розпадом і утворенням порожнин у центрі вогнища. Зміни в легеневій тканині у хворих на лімфому Ходжкіна можуть поєднуватися зі збільшеними лімфатичними вузлами середостіння та з ураженням плеври.
З органів найчастіше уражаються легені — 20–30% випадків. Ураження кісток виникає в 14–20%, в основному локалізується у хребцях, ребрах, кістках таза. При лімфомі Ходжкіна лише у 10–12% відзначають ураження кісткового мозку. В мієлограмі може реєструватися мієлоїдна та мегакаріоцитарна гіперплазія. При лімфомі Ходжкіна ІV стадії та ураженні кісток у кістковому мозку можуть виявлятися поодинокі клітини Березовського — Штернберга.
КЛАСИФІКАЦІЯ
Гістологічно виділяють класичну лімфому Ходжкіна, яка відзначається у 95% випадків, із 4 гістологічними підтипами (нодулярний склероз, багатий лімфоцитами, змішано-клітинний тип, лімфоїдного виснаження) та лімфому Ходжкіна, лімфоїдне переважання, нодулярний варіант.
Лімфома Ходжкіна, лімфоїдне переважання, нодулярний варіант, розглядається окремо, оскільки за своїми клінічними особливостями (млявий персистуючий перебіг), морфологічними відмінностями (наявність діагностичних крупних L- і H-клітин на тлі переважання лімфоїдних елементів, тоді як класичні клітини Рід — Штернберга виявляють вкрай рідко) та імунофенотиповими ознаками (CD20+, CD79a+, CD15–, CD30–) суттєво відрізняється від класичної форми лімфоми Ходжкіна та потребує окремих підходів у лікуванні.
ДІАГНОСТИКА
Основним методом діагностики лімфоми Ходжкіна є гістологічний. Для отримання матеріалу рекомендують проведення ексцизійної біопсії пухлини (здебільшого лімфатичного вузла) чи трепанобіопсії вогнища ураження (частіше виконується при ізольованому ураженні середостіння та за відсутності збільшених периферичних лімфатичних вузлів) з подальшим його гістологічним дослідженням, де проводять пошук, насамперед, гігантських клітин Рід — Штернберга та клітин Ходжкіна (рис. 18.4).
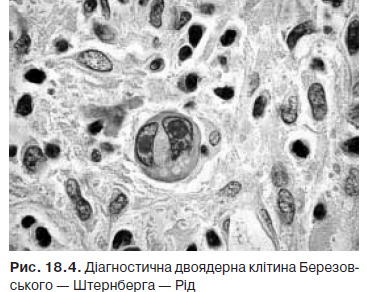
Імуногістохімічне дослідження матеріалу біопсії при лімфомі Ходжкіна проводиться за показаннями (мінімальна імуногістохімічна панель: для класичних варіантів — CD3, CD15, CD20, CD30, CD45, для варіанта лімфоїдного переважання — CD3, CD15, CD20, CD21, CD30, CD57).
Після встановлення діагнозу лімфоми Ходжкіна виконується низка обстежень для встановлення стадії захворювання та визначення групи прогностичного ризику, що має вирішальне значення для вибору тактики і схем лікування.
Клінічне стадіювання лімфоми Ходжкіна, що враховує, насамперед, розповсюдження процесу по обидва боки діафрагми та наявність чи відсутність системних симптомів, проводиться за системою Ann Arbor, модифікованою за Cotswolds у 1989 р.
Клінічне стадіювання, окрім об’єктивного огляду хворого, базується на виконанні таких процедур:
- КТ шиї, грудної клітки, черевної порожнини, малого таза із внутрішньовенним контрастуванням (за неможливості виконання КТ: рентгенологiчне дослiдження органiв грудної клiтки, УЗД органів черевної порожнини, малого таза);
- трепанобіопсія кісткового мозку з подальшим гістологічним дослідженням.
КТ шиї, органів грудної та черевної порожнини, а також малого таза є обов’язковим інструментальним методом дослідження хворих, який застосовують для з’ясування поширеності патологічного процесу як при первинному ураженні, так і при оцінці відповіді на лікування, а також при верифікації рецидивів захворювання.
ПЕТ — це сучасний метод молекулярної діагностичної візуалізації з використанням внутрішньовенно контрасту 18F-фтордезоксиглюкозою (18F-ФДГ). Останні роки цей метод широко використовують у клінічній онкології. Знання переваг та недоліків методу, особливостей його використання у хворих різних нозологічних груп дозволяють суттєво підвищити точність отриманих результатів. У комплексі з традиційними методами структурної візуалізації (УЗД, КТ, МРТ) ПЕТ дозволяє уточнити стадію захворювання (метод надає можливість оцінити стан як лімфатичних вузлів, так і виявити вогнища екстранодального ураження). Особливо велике значення має ПЕТ для діагностики вогнищевого ураження селезінки, дифузного ураження печінки та селезінки, ураження скелета, кісткового мозку. Точність ПЕТ у визначенні стадії захворювання перевищує 95%, тоді як при використанні методів структурної візуалізації вона становить біля 60–70%. Використання ПЕТ у процесі лікування хворих призводить до уточнення стадії захворювання у >30% хворих. ПЕТ суттєво переважає КТ у точності оцінки ефективності проведеного лікування, оскільки надає можливість виявити в резидуальній пухлинній масі залишки життєздатної пухлинної тканини. ПЕТ може бути методом ранньої оцінки відповіді на проведене лікування. Результати ПЕТ, виконаної вже після першого курсу хіміотерапії, дозволяють не лише з високою точністю виявити ефективність лікування у конкретного пацієнта, але має більш прогностичне значення у 90–95%.
Використання сучасної гібридної технології ПЕТ/КТ, яка надає можливість при одному дослідженні зіставити структурні та фізіологічні дані, суттєво підвищує інформативність ПЕТ та відкриває нові направлення використання методу у хворих на лімфоми (наприклад для планування ПТ).
Характерних змін у периферичній крові на ранніх стадіях захворювання не існує. При пізніх стадіях у хворих відзначається підвищення швидкості осідання еритроцитів (ШОЕ), нейтрофільний лейкоцитоз зі зсувом лейкоцитарної формули вліво, відносна чи абсолютна лімфопенія (внаслідок ураження пухлинним процесом лімфоїдної тканини), еозинофілія, моноцитоз, анемія.
У біохімічному аналізі крові необхідно визначити вміст альбуміну (рівень якого належить до факторів ризику) та сечовини при великій пухлинній масі.
Стадіювання лімфом здійснюється за системою Ann Arbor у модифікації Costwold:
Стадія І — ураження однієї лімфатичної зони або структури чи локалізоване ураження одного екстралімфатичного органа або тканини (1Е) у межах одного сегмента.
Стадія ІІ — ураження двох або більше лімфатичних зон по один бік діафрагми (наприклад середостіння — одна зона, кореня легень — окремі від середостіння самостійні зони) або локалізоване ураження одного екстралімфатичного органа чи тканини та їх регіонарних лімфатичних вузлів з ураженням інших лімфатичних зон по той же бік діафрагми або без нього. Для ІІ стадії необхідно зазначити кількість уражених лімфатичних зон, наприклад 2.
Стадія ІІІ — ураження лімфатичних вузлів або структур з обох боків діафрагми, що може поєднуватися з локалізованим ураженням одного екстралімфатичного органа чи тканини (3Е) або з ураженням селезінки (S), або з ураженням того й іншого (3Е + S). Рекомендується виділяти верхні абдомінальні лімфатичні вузли (ворота печінки, селезінка) — стадія ІІІ1 і нижні абдомінальні (парааортальні, мезентеріальні) стадія ІІІ2.
Стадія IV — дисеміноване ураження одного або декількох екстралімфатичних органів з ураженням лімфатичних вузлів чи без них, або ізольоване ураження екстралімфатичного органа з ураженням окремих лімфатичних вузлів. При ураженні печінки чи кісткового мозку — завжди встановлюється IV стадія захворювання.
Буквами «S» позначають ураження селезінки; «Е» — екстранодальне ураження у межах одного сегмента; «Х» — масивне ураження лімфатичних вузлів.
Окремо виділяють симптоми інтоксикації: «В» — наявність одного або більше таких клінічних симптомів, як прогресуюча втрата маси тіла на 10% за останні 6 міс, профузне пітніння, підвищення температури тіла >38 °С без наявності інфекційного захворювання чи інших причин; «А» — відсутність вищезазначених симптомів.
Після отримання результатів гістологічного дослідження та визначення стадії захворювання необхідно визначити фактори несприятливого прогнозу перебігу захворювання.
Основні фактори ризику несприятливого прогнозу при лімфомі Ходжкіна І–ІІ стадії (GHSG):
- Наявність масивного >10 см (bulky disease) ураження лімфатичних вузлів;
- Підвищення ШОЕ (≥50 мм/год за відсутності В-симптомів та ≥30 мм/год за наявності В-симптомів);
- Наявність ≥3 зон ураження;
- Наявність В-симптомів;
- Наявність ≥2 екстранодальних вогнищ ураження;
Несприятливі прогностичні фактори при лімфомі Ходжкіна III–IV стадії такі:
- Вік ≥45 років
- Чоловіча стать;
- IV стадія патологічного процесу;
- Рівень гемоглобіну <105 г/л;
- Кількість лейкоцитів ≥15,0 · 109/л;
- Лімфоцитопенія <600/мкл або <8%;
- Вміст альбуміну <40,0 г/л.
Згідно з сучасними рекомендаціями, побудованими на обґрунтованих висновках рандомізованих клінічних досліджень кооперативних груп, розрізняють такі прогностичні групи пацієнтів із лімфомою Ходжкіна (табл. 18.1).
Прогностичні групи пацієнтів із лімфомою Ходжкіна
Ступінь ризику | Стадія |
Низький | І/ІІ без факторів ризику |
Проміжний | І–ІІА з ≥1 факторами ризику |
Високий | ІІВ з ≥1 факторами ризику, |
ДИФЕРЕНЦІЙНИЙ ДІАГНОЗ
Диференційну діагностику лімфоми Ходжкіна необхідно проводити, перш за все, із захворюваннями, які супроводжуються збільшенням лімфатичних вузлів. До них належать хронічний лімфолейкоз, туберкульоз лімфатичних вузлів, неходжкінська злоякісна лімфома, саркоїдоз Беньє — Бекка — Шауманна, метастази раку в лімфатичних вузлах, хвороба Бриля — Симерса, інфекційний мононуклеоз, ВІЛ. Найскладніший диференційний діагноз із неходжкінськими лімфомами, при яких основним симптомом також є збільшення лімфатичних вузлів та інших лімфоїдних органів. Відзначимо, що для неходжкінської лімфоми нехарактерне злиття лімфовузлів у конгломерати. Генералізована лімфаденопатія, ураження кільця Вальдеєра на початку захворювання рідко відмічається при лімфомі Ходжкіна, частіше — при неходжкінській лімфомі. Також часто при індолентній неходжкінській лімфомі виявляють лімфоцитоз у периферичній крові та в кістковому мозку. Вирішальне значення у встановленні точного діагнозу має біопсія лімфатичних вузлів (визначення клітин Березовського — Штернберга). При імунофенотипуванні експресія загального антигену лейкоцитів (СD45) у гістологічному препараті при лімфомі Ходжкіна в більшості випадків відсутня, що дозволяє відрізнити його від неходжкінської лімфоми.
ЛІКУВАННЯ
Лікування хворих на лімфому Ходжкіна необхідно проводити у спеціалізованих закладах. Основними методами лікування є медикаментозний та променевий. Хірургічне втручання використовують лише з метою діагностики та у разі ускладнень основного захворювання. Численні дослідження підтверджують переваги застосування комбінованого підходу в лікуванні пацієнтів із лімфомою Ходжкіна всіх стадій (табл. 18.2–18.3).
Алгоритм лікування класичної лімфоми Ходжкіна
Стадія захворювання | Обсяг стандартного лікування |
І/ІІ без факторів ризику (низький ступінь ризику), вік хворого <60 років | 2–4 курси ПХТ за схемою АВVD + ПТ на уражені зони, сумарна вогнищева доза (СВД) 20–30 Гр |
І–ІІА з ≥1 факторами ризику (проміжний ступінь ризику), вік хворого <60 років | 4 курси ПХТ за схемою ABVD або 2 курси ПХТ за схемою BEACOPP -esc та 2 курси ПХТ за схемою ABVD + ПТ на уражені зони СВД 30 Гр |
ІІВ з ≥1 факторами ризику, ІІІ–IV стадія (високий ступінь ризику), вік хворого <60 років | 6–8 курсів ПХТ за інтермітуючими або багатокомпонентними режимами (за схемою АВVD, ВЕАСОРР-14 або ВЕАСОРР-esc) ± ПТ на залишкову пухлину СВД 30 Гр; можлива зміна терапії з ініціальної схеми ВЕАСОРР-14 або ВЕАСОРР-esc на ВЕАСОРР при досягненні повної відповіді, часткової відповіді або при вираженій гематологічній токсичності. У разі недостатньої відповіді на лікування при проміжній оцінці відповіді (після 3–4 циклів) можна передбачити зміну терапії зі схеми ABVD на режими ВЕАСОРР-14 та ВЕАСОРР-esc або при прогресуванні хвороби salvage-терапія |
I–IV, вік хворого >60 років, або хворі з наявністю тяжкої супутньої патології | 2–8 курсів за схемами АВVD, АВVD/СОРР, за схемою АВV/СОРР ± ПТ на залишкову пухлину + ПТ СВД 30 Гр |
Алгоритм лікування лімфоми Ходжкіна, лімфоїдне переважання, нодулярний варіант
Стадія захворювання | Обсяг стандартного лікування |
Стадії IА без факторів ризику | ПТ на уражену зону 30 Гр |
Усі інші стадії лікуються як класична лімфома Ходжкіна ± ритуксимаб | |
Обов’язковою вимогою при лікуванні хворих на лімфому Ходжкіна є проміжна оцінка відповіді на терапію. Рекомендується проводити оцінку відповіді на терапію після 2–4 циклів ПХТ та після завершення останнього циклу за допомогою тих самих методів діагностики, що застосовували до лікування (КТ, УЗД, рентгенографія). Загальновизнаною системою оцінки відповіді на лікування лімфоми Ходжкіна є рекомендації Міжнародної робочої групи (Сheson В., 1999; 2007) (табл. 18.4).
Критерії відповіді на терапію у пацієнтів зі злоякісними лімфомами, згідно з рекомендаціям Міжнародної групи з дослідження неходжкінської злоякісної лімфоми (СHESON, 1999)
Категорія відповіді на терапію | Суб’єктивні критерії | Лімфатичні вузли | Печінка, селезінка | Кістковий мозок |
Повна відповідь (ПВ) | Повне зникнення усіх клінічних ознак та симптомів | Нормалізація розмірів лімфовузлів Якщо розміри лімфовузлів до початку терапії становили >1,5 см у діаметрі, то необхідною умовою є зменшення їх до 1,5 см. Якщо розміри уражених лімфовузлів становили 1,1–1,5 см, то необхідною умовою є зменшення їх до <1 см | НОРМА Якщо в момент встановлення діагнозу було зареєстровано збільшення селезінки, то при встановленні ПВ необхідно отримати дані про зменшення її розмірів, а також відсутність даних про збільшення при пальпації | НОРМА Відсутність ураження кісткового мозку за даними аспірата та біопсії, що виконуються в тих самих місцях, як і при першому дослідженні |
Повна відповідь непідтверджена (ПВн) | Повне зникнення усіх клінічних ознак та симптомів | Розмір резидуальних лімфовузлів >1,5 см за максимальним розміром або зменшення >75% | НОРМА Якщо в момент встановлення діагнозу зареєстровано збільшення селезінки, то при встановленні ПВ необхідно отримати дані про зменшення її розмірів | Відсутність ураження кісткового мозку за даними аспірата та біопсії, що виконуються в тих самих місцях, як і при першому дослідженні |
Часткова відповідь (ЧВ) | Зникнення усіх клінічних ознак та симптомів | Зменшення розмірів на >50% | Зменшення у розмірах печінки та селезінки | Уражений або не оцінювався |
Стабілізація захворювання (СЗ) | Немає симптомів прогресування | Без змін | Без змін | Без змін |
Рецидив (визначається для пацієнтів, у яких було досягнуто ПВ або ПВн) | Клінічні ознаки захворювання | Ураження нових або збільшення на >50% старих вогнищ ураження | Збільшення у розмірі | Ураження |
Прогресування (визначається для пацієнтів, у яких було досягнуто ЧВ або СЗ) | Клінічні ознаки захворювання | Ураження нових або збільшення на >50% старих вогнищ ураження | Збільшення у розмірі | Ураження |
При лікуванні у 80–95% випадків у хворих на лімфому Ходжкіна можна досягти позитивної відповіді на лікування. Це залежить від стадії патологічного процесу та факторів прогнозу (при більш ранніх стадіях та відсутності несприятливих факторів — 95%, при пізніх стадіях із наявністю несприятливих факторів — 80%). Проте кількість терапевтичних невдач (первинно-рефрактерний перебіг лімфоми чи виникнення рецидиву) залишається суттєвою проблемою лікування хворих на лімфому Ходжкіна.
При використанні сучасних ризикадаптованих підходів терапії у 10–15% випадків лімфоми Ходжкіна виявляються рефрактерними до терапії першої лінії. До 30% пацієнтів мають рецидив захворювання, який у 90% випадків виникає впродовж перших двох років. Стандартна хіміотерапія другої лінії дозволяє досягнути у них тривалої безрецидивної виживаності лише у 10–15%.
Застосування хіміопрепаратів у стандартних дозах при лікуванні первинно-рефрактерного перебігу лімфоми Ходжкіна або при рецидиві не має терапевтичного ефекту.
Зважаючи на відомий для багатьох цитостатичних препаратів дозозалежний протипухлинний ефект, можлива ескалація їх дози. Але це значно підвищує органну токсичність, перш за все — гематологічну (мієлотоксичність), яка навіть при проведенні стандартної хіміотерапії становить 40–90% і може спричиняти тяжкі ускладнення терапії (анемію, кровотечі внаслідок тромбоцитопенії, тяжкі інфекції при нейтропенії).
Подолати ці проблеми дозволяє введення аутологічних стовбурових клітин (СК), після чого відбувається відновлення нормального трьох-паросткового гемопоезу.
Перші трансплантації гемопоетичних стовбурових клітин (ТГСК) були проведені групами G. Mathe та E.D. Thomas і R. Storb у другій половині 1960-х років. Сьогодні ТГСК успішно використовуються при багатьох захворюваннях і є одним із стандартних методів терапії лімфом. У країнах Європейського Союзу загалом виконують >25 тис. трансплантацій на рік, з них майже половина — ауто-ТГСК при лімфопроліферативних захворюваннях.
ТГСК передбачає проведення високодозової (ВДХТ), тобто мієлоаблятивної хіміотерапії, після чого вводять трансплантат — СК. Залежно від походження СК трансплантацію поділяють на алогенну (від іншої людини-донора) та аутологічну, коли використовуються власні клітини пацієнта. При алогенній ТГСК, окрім протипухлинної дії ВДХТ, реалізується терапевтичний ефект «трансплантат проти пухлини». При ауто-ТГСК протипухлинний ефект зумовлює лише ВДХТ, а введення гемопоетичних СК має на меті лише відновлення кровотворення.
Загальний план проведення ВДХТ включає декілька етапів. Спочатку проводять курси ПХТ другої лінії, спрямовані на зменшення пухлинного об’єму перед ВДХТ та визначення хіміочутливості пухлини до терапії. На сьогодні найпоширенішими є схеми ПХТ другої лінії DHAP, ESHAP, MINE, ICE, GVP, IVEG та ін.
Для проведення ТГСК потрібно зібрати та зберегти СК. При лікуванні лімфом у більшості випадків для цього використовують СК, отримані з периферичної крові. Ідентифікацію та підрахунок СК проводять декількома методами, найпоширенішим із них є визначення кількості CD34+-клітин. Процедура колекції проводиться після відновлення гемопоезу після хіміотерапії з використанням гранулоцитарного чи гранулоцитарно-макрофагального колонієстимулюючого фактора. Після застосування колонієстимулюючого фактора кількість СК у периферичній крові зростає від 0,1–1 до 10 CD34+-клітини в 1 мкл. Колекція гемопоетичних СК проводиться методом безперервного цитоаферезу периферичної крові з використанням клітинних сепараторів. Для відновлення гемопоезу після ВДХТ мінімальною є доза гемопоетичних СК 2 · 106 CD34+-клітин на 1 кг маси тіла пацієнта. Після отримання гемопоетичних СК необхідне проведення їх кріоконсервування, що дозволяє тривале зберігання при температурі не вище –80 °С без втрати життєдіяльності. Оптимальним є метод зберігання СК у парах рідкого азоту.
Проведення ВДХТ перед ТГСК називається кондиціонуванням. Найчастіше це курс багатокомпонентної хіміотерапії. При лімфомі Ходжкіна використовують кондиціонування за схемами BEAM, CBV, ICE. Після проведення ПХТ через 24–72 год пацієнту вводять гемопоетичні СК, які попередньо розморожують, що є власне процесом трансплантації. Відновлення нормального трьохпаросткового кровотворення частіше відбувається на 10–14-й день після аутотрансплантації.
При лімфомі Ходжкіна проведення ВДХТ з аутологічною ТГСК є найбільш ефективною терапією при лікуванні первинно-рефрактерних форм захворювання та рецидивів. Проводяться дослідження ефективності алогенної ТГСК, отримано позитивні результати, але остаточна роль цього методу при несприятливому перебігу лімфоми Ходжкіна залишається експериментальним підходом.
НЕХОДЖКІНСЬКІ ЛІМФОМИ
Неходжкінські лімфоми — група лімфопроліферативних пухлин, які відрізняються за біологічними властивостями, морфологічною будовою, клінічними проявами, відповіддю на терапію та прогнозом. Одні види лімфом мають повільний перебіг та сприятливий прогноз і інколи тривалий час не потребують спеціального лікування. Такі лімфоми називають індолентними. Ряд інших лімфом, навпаки, характеризуються швидким прогресуванням, вираженими клінічними проявами і потребують негайного початку терапії. Такі лімфоми називають агресивними.
ЕТІОЛОГІЯ
Етіологію лімфом досі не встановлено. Розглядається ряд факторів, що можуть підвищити ризик розвитку неходжкінської лімфоми, серед яких інфекційні агенти, іонізуюче випромінення, імунодефіцит та інші фактори. Встановлена роль вірусу Епштейна — Барр у розвитку класичної лімфоми Беркіта, розповсюдженої в ендемічних малярійних регіонах Африки. Загалом ДНК вірусу Епштейна — Барр виявляється у 10–30% випадків лімфом. Таким чином, вірус Епштейна — Барр може бути як безпосередньою причиною розвитку деяких варіантів, так і відображати наявність імунної дисфункції при неходжкінській лімфомі. Ризик розвитку неходжкінської лімфоми також суттєво підвищується за наявності у хворого ВІЛ. Також доведена роль Chlamydia psittaci у виникненні МALT-лімфоми з ураженням ока та Helicobacter pylorі при MALT-лімфомі шлунка. Роль пестицидів у розвитку неходжкінської лімфоми підтверджується тим фактом, що при довготривалому контакті з ними захворюваність на неходжкінську лімфому підвищується у 2–8 разів. Існують дані щодо ролі іонізуючого випромінення в розвитку неходжкінської лімфоми, застосування хіміотерапії також призводить до підвищення ризику розвитку цієї групи захворювань. Встановлений зв’язок між наявністю імунної дисфункції та частотою розвитку неходжкінської лімфоми при ряді спадкових імунодефіцитних синдромів: синдромі Віскотта — Олдрича, Чедіака — Хігаші, атаксії — телеангіектазії. Достовірно відомо, що імуносупресивна терапія при проведенні трансплантації органів і тканин призводить до підвищення частоти захворюваності на неходжкінську лімфому в декілька десятків разів.
КЛІНІЧНА КАРТИНА
В основному клінічна картина залежить від локалізації вогнищ ураження та пов’язаних із ними порушень функцій різних органів. Так, при локалізації процесу в середостінні з’являються симптоми здавлення верхньої порожнистої вени, розвивається задишка, ціаноз, набряк обличчя та шиї. Збільшення мезентеріальних або заочеревинних лімфатичних вузлів може призвести до розвитку асциту і непрохідності кишечнику; при здавленні загальної жовчної протоки лімфатичними вузлами у воротах печінки розвивається механічна жовтяниця; при локалізації лімфатичних вузлів поблизу хребта з’являються корінцеві симптоми і навіть параплегії внаслідок проникнення пухлини в хребці та здавлення спинного мозку. Крім лімфатичних вузлів, у специфічний процес нерідко залучаються органи і тканини — печінка, селезінка, мигдалики, носова частина глотки, шлунок, кишки, плевра, легені, кістки, шкіра, м’які тканини, кістковий мозок та ін. При ураженні легень з’являються симптоми, що нагадують рак легені: нав’язливий кашель, задишка, біль у грудях, ателектаз, плевральний випіт, іноді геморагічного характеру.
Безболісне збільшення периферичних лімфовузлів — основна скарга хворих на неходжкінські лімфоми. Звертають увагу на розміри лімфовузлів, консистенцію і болючість, а також на кількість лімфовузлів, що пальпуються. Неходжкінській лімфомі властива щільноеластична консистенція лімфатичних вузлів.
В-симптоми (прогресуюча втрата маси тіла на 10% за останні 6 міс, профузне пітніння, підвищення температури тіла >38 °С без наявності інфекційного захворювання чи інших причин) характерні, але не специфічні лише для неходжкінських лімфом симптоми. Можливі також скарги на загальну слабкість і виражену втомлюваність, невідповідні глибині анемії.
Неприємні відчуття і біль у животі бувають наслідком спленомегалії, гідронефрозу або здавлення кишечнику лімфатичними вузлами. Біль у кістках свідчить про їх вогнищеве ураження або про дифузне ураження кісткового мозку. Нейрогенний біль може бути спричинений здавленням спинного мозку, нервових сплетень, нейролейкозом. Біль у спині може бути зумовлений також ураженням заочеревинних лімфовузлів, часто з проростанням у великий поперековий м’яз.
КЛАСИФІКАЦІЯ
Сучасну класифікацію неходжкінської лімфоми проводять згідно з рекомендаціями ВООЗ, 2008. Виділення варіантів лімфом за типом перебігу (агресивні, індолентні) відбувається за класифікацією REAL (табл. 18.5).
Таблиця 18.5
Європейсько-американська класифікація лімфом (REAL)
В-клітинні лімфоми | Т-клітинні лімфоми |
Індолентні | |
Хронічна лімфолейкемія Лімфома з малих лімфоцитів Імуноцитома, лімфоплазмоцитарна лімфома Волосатоклітинна лейкемія Плазмоцитома (мієлома) Лімфома маргінальної зони селезінки Екстранодальна лімфома маргінальної зони Нодулярна лімфома із клітин маргінальної зони Лімфома із клітин фолікулярного центра Лімфома із клітин мантії | Хронічна лімфоцитарна лейкемія Лейкемія з великих грануловмісних лімфоцитів Грибоподібний мікоз |
Агресивні лімфоми | |
Дифузна великоклітинна лімфома | Анапластична великоклітинна лімфома Периферична Т-клітинна лімфома |
Високоагресивні лімфоми | |
Прекурсорна В-лімфобластна лейкемія/лімфома Лімфома Беркіта | Прекурсорна Т-лімфобластна лейкемія/лімфома Т-клітинна лімфома/лейкемія дорослих (HTLV+) |
ДІАГНОСТИКА
Діагностичний пошук неходжкінських лімфом має включати об’єктивне обстеження хворого та лабораторно-інструментальні методи дослідження, в тому числі інвазивні.
При діагностиці неходжкінських лімфом застосовують такі ж методи, як і при лімфомі Ходжкіна.
Імуногістохімічне дослідження матеріалу біопсії при неходжкінських лімфомах є обов’язковим методом дослідження. За допомогою імунофенотипування проводять визначення диференціювальних антигенів (CD-антиген) на поверхні та у цитоплазмі пухлинних клітин. Це дослідження особливо важливе для уточнення варіанта клітинного походження лімфопроліферативних захворювань (В- або Т-лінія), а також стадії диференціювання та ступеня зрілості клітин. Імунофенотипові особливості різних морфологічних варіантів неходжкінської лімфоми є високоінформативними для діагностики та прогнозу захворювання. Нижченаведено імунофенотипи В- та Т-клітинних пухлин.
- Фолікулярна лімфома: CD10+, bcl-2+, CD23+/–, CD43–, CD5–, CD20+, cyclin D1–
- Неходжкінська лімфома маргінальної зони: CD10–, CD5–, CD20+, CD23–/+, CD43–/+, cyclin D1–, bcl-2
- Мантійноклітинна лімфома: CD5+, CD20+, CD43+, CD23–/+, cyclin D1+, CD10–/+
- Дифузна В-великоклітинна лімфома: CD20+, CD45+, CD3–
- Лімфома Беркіта: sIg+, CD10+, CD20+, TdT–, Ki67+ (100%), bcl-2–, bcl-6+
- Естранодальна Т-/NK-клітинна лімфома, назальний тип: CD3–/+, CD5+, CD7–/+, CD30–/+
- Грибоподібний мікоз: CD3+, CD4+, CD5+, CD8–/+
- Ангіоімунобластна лімфома: CD3+, CD4–/+, CD5+, CD7+, CD8–/+ CD30–/+
При неходжкінських лімфомах в загальному аналізі крові може відмічатися лейкоцитоз, лімфоцитоз, нейтропенія, анемія, ШОЕ, тромбоцитопенія.
У біохімічному аналізі крові необхідно визначити активність ферменту лактатдегідрогенази (підвищений рівень свідчить про наявність активного пухлинного процесу в організмі та є фактором ризику) і сечовини при великій пухлинній масі.
Такі показники, як рівень білірубіну, АлАТ, АсАТ, креатиніну, нададуть інформацію стосовно залучення в патологічний процес печінки або про наявність супутньої патології, що необхідно для прийняття рішення про можливість проведення специфічного лікування.
Цитологічне та/чи гістологічне дослідження кісткового мозку відіграє важливу роль у діагностиці неходжкінської лімфоми та є одним із важливих критеріїв проведення диференційного діагнозу лейкемоїдних реакцій та поширення злоякісного процесу.
Верифікація неходжкінських лімфом неможлива без застосування таких методів, як імуногістохімічний та цитогенетичний, а також метод імунофенотипування клітин периферичної крові та кісткового мозку.
Ендоскопію чи колоноскопію проводять при ураженні лімфовузлів шлунково-кишкового тракту при неходжкінських лімфомах, а також за наявності симптомів ураження шлунково-кишкового тракту.
При особливих клінічних варіантах неходжкінської лімфоми додатково застосовують: КТ головного мозку, люмбальну пункцію, ФГДС з біопсією, остеосцинтиграфію та МРТ.
Після встановлення діагнозу на підставі клінічних та морфологічних ознак визначають поширеність патологічного процесу відповідно до Ann Arbor-класифікації (1971), модифікованої за Cotswolds (1973) (див. Лімфома Ходжкіна).
Після отримання усіх результатів досліджень для вирішення подальшої тактики лікування хворого необхідним є визначення факторів несприятливого прогнозу перебігу захворювання. Загальноприйнятою прогностичною системою є міжнародний прогностичний індекс (табл. 18.6).
Міжнародний прогностичний індекс для неходжкінських лімфом (IPI)
Прогностичний фактор виживання | Міжнародний прогностичний індекс | |||
Критерій | 0 балів | 1 бал | Група ризику | Кількість балів |
Вік, років | <60 | >60 | ||
N; <N | >N | Низького | 0; 1 | |
Загальний стан (за ECOG) | 0; 1 | 2; 3; 4 | Низького проміжного | 2 |
Стадія за Ann Arbor | I/II | III/IV | Високого проміжного | 3 |
Екстранодальні | 1; <1 | >1 | Високого | 4; 5 |
Окремо виділяють міжнародний прогностичний індекс для фолікулярних лімфом (FLIPI) (табл. 18.7).
Міжнародний прогностичний індекс для фолікулярних лімфом (FLIPI)
Прогностичний фактор виживання | Міжнародний прогностичний індекс | |||
Критерій | 0 балів | 1 бал | Категорія | Кількість балів |
Вік | <60 років | >60 років | ||
N; <N | >N | Низький | 0; 1 | |
Рівень гемоглобіну | >120 | <120 | Проміжний | 2 |
Стадія за Ann Arbor | I/II | III/IV | Високий | ≥3 |
Кількість нодальних зон ураження | < 4 | >4 | ||
Лікування агресивних неходжкінських лімфом
Лікування хворих на неходжкінську лімфому необхідно проводити у спеціалізованих закладах. Основними методами лікування є медикаментозний та променевий (табл. 18.8).
Алгоритм лікування хворих на агресивні неходжкінські лімфоми
Стадії та прогностичні фактори | Обсяг лікування |
I, ІІ стадія без негативних прогностичних факторів, пухлина <10 см |
|
I, ІІ стадія з негативними прогностичними факторами (лактатдегідрогеназа, ІІ стадія, вік >60 років, статус ЕСОG >2), пухлина <10 см |
|
І, II стадія, пухлина >10 см |
|
IІІ–IV стадія |
|
Лікування індолентних неходжкінських лімфом
Існує декілька підходів до лікування індолентних лімфом (табл. 18.9):
- чекати та спостерігати;
- монотерапія алкілуючими препаратами (хлорамбуцил);
- комбінована ПХТ (COP, CVP, СНОР);
- комбінована ПХТ із застосуванням пуринових аналогів (FC, FMD);
- терапія моноклональними антитілами (ритуксимаб);
- ПХТ + моноклональні антитіла (R-Clb, R-COP, R-CVP, R-CHOP, R-FC, R-FMD);
- ВДХТ + трансплантація СК (при рефрактерних та рецидивуючих лімфомах).
Алгоритм лікування хворих на індолентні неходжкінські лімфоми
Стадія | Обсяг лікування хворих на індолентні неходжкінські лімфоми |
I, ІІ |
|
IІІ–IV |
|
Показання для початку терапії індолентних лімфом:
- Симптоми хвороби
- Порушення функції органів
- Постійне прогресування хвороби
- Нодальні або екстранодальні ураження >7 см (bulky)
- Вторинна цитопенія, пов’язана з лімфомою
Після проведення трьох курсів ПХТ та завершення лікування обов’язковою є оцінка відповіді на терапію, яку виконують за допомогою тих самих методів діагностики, що застосовували до лікування (КТ, УЗД, рентгенографія, дослідження кісткового мозку). Загальновизнаною системою оцінки відповіді на лікування неходжкінської лімфоми є рекомендації Міжнародної робочої групи (Сheson В., 1999; 2007) (див. Лімфома Ходжкіна).
При первинному лікуванні неходжкінської лімфоми рецидив чи первинно-рефрактерний перебіг розвивається у 50% хворих, залежно від варіанта лімфоми, прогностичної групи. Для цієї групи пацієнтів оптимальним підходом є проведення хіміотерапії за схемами другої лінії, після чого бажане проведення ВДХТ. Принципи цієї методики аналогічні таким при лімфомі Ходжкіна і викладені у відповідному розділі.
Зважаючи на те, що група неходжкінської лімфоми гетерогенна за перебігом та прогнозом, показання до ВДХТ відрізняються для різних видів лімфом. На сьогодні ВДХТ є стандартною терапією при таких клінічних випадках:
- дифузна В-великоклітинна лімфома: рецидив чи рефрактерний перебіг;
- лімфома із клітин мантійної зони: консолідація першої ремісії, хіміочутливий рецидив;
- фолікулярна лімфома: відсутність відповіді на ініціальну терапію, ранній рецидив, другий та наступний рецидив, трансформація у дифузну В-великоклітинну лімфому;
- Т-клітинні ALK-негативні лімфоми: консолідація в І ремісії, первинно-рефрактерний перебіг (за умови відповіді на терапію другої лінії).
ХРОНІЧНА ЛІМФОЇДНА ЛЕЙКЕМІЯ
Хронічна лімфоїдна лейкемія (ХЛЛ; Chronic lymphocytic leukemia) — індолентна лімфоїдна пухлина (Low-Grade) В-клітинного походження, при якій відбувається злоякісна проліферація малих, морфологічно зрілих В-лімфоцитів із накопиченням у кістковому мозку, периферичній крові та лімфоїдних органах.
Захворюваність на ХЛЛ за оцінками різних авторів становить від 2,5 до 4,5 на 100 тис. населення на рік у осіб віком до 60 років. У людей віком старше 60 років захворюваність підвищується до 20 випадків на 100 тис. населення на рік. Чоловіки хворіють частіше за жінок у співвідношенні 2:1. За даними Національного канцер-реєстру в Україні захворюваність на ХЛЛ становить 3,53 випадка на 100 тис. населення (абсолютна кількість — 1426) на рік, поширеність — 22,19 випадка на 100 тис. населення (абсолютна кількість — 8966).
ЕТІОЛОГІЯ
Етіологія хронічної лімфоцитарної лейкемії, як і всіх гемобластозів, до кінця не вивчена. Відома роль деяких мутагенних факторів у виникненні хронічних лейкозів (хімічні мутагенні фактори, опромінення, віруси). Широко обговорюється вірусна теорія. Доведено, що хронічний лімфолейкоз у мишей викликають вірус Гроса, у щурів — вірус Молоні, Шварца — Скульмана. У 1982 р. виділений ретровірус від хворого на Т-клітинний лейкоз — людський Т-клітинний вірус 1 (НТLV-1). Встановлено, що вірус за допомогою реверсивної транскриптази впроваджується в ДНК клітини господаря, змінює генетичний код клітини, відбувається мутація, клітина стає злоякісною. На сьогодні показано, що на відміну від інших лейкозів, ХЛЛ не індукується зовнішніми чинниками, такими як іонізуюча радіація. Описано спадковий генез деяких випадків ХЛЛ, роль етнічних факторів. Відомі як домінантні, так і рецесивні успадковані випадки. Описано випадки ХЛЛ у декількох членів однієї родини. Існують численні спадкові захворювання, пов’язані з дефектом імунітету — хвороба Луї — Барр, хвороба Братона, які призводять до виникнення ХЛЛ. Існує ряд етнічних чинників у виникненні ХЛЛ: у США висока захворюваність відзначена серед осіб єврейської національності. Казахи хворіють на ХЛЛ істотно менше порівняно з населенням Росії. В Японії захворюваність на хронічну лімфоцитарну лейкемію також низька. При ХЛЛ часто виявляють хромосомні аномалії, частіше в 14, 11, 3, 18-й парах хромосом.
ПАТОГЕНЕЗ
Розвиток ХЛЛ відбувається в результаті мутації однієї клітини — частіше клітини — попередника лімфопоезу (В- або Т-клітинного походження). Виникнення пухлини починається в кістковому мозку. На відміну від гострого лейкозу, при ХЛЛ кожна рання пухлинна клітина здатна, крім некерованого розмноження, до подальшого дозрівання, диференціювання до зрілих лімфоцитів. Пухлинні лімфоцити несуть на поверхні Ig одного класу і підкласу. З розвитком захворювання в кістковому мозку прогресує лімфоїдна проліферація, пригнічується нормальне кровотворення. Відбувається дисемінація пухлинних клітин із кісткового мозку у кровотворні органи та інші системи з подальшим розростанням лімфоїдної тканини в лімфовузлах, печінці, селезінці. У периферичній крові наростає лімфоцитарний лейкоцитоз. Генез цитопенії при ХЛЛ може також носити частіше аутоімунний характер, з’являються ознаки аутоімунної гемолітичної анемії, тромбоцитопенії.
КЛІНІЧНА КАРТИНА
У 25% пацієнтів захворювання перебігає безсимптомно і виявляється випадково під час медичного огляду чи лабораторних досліджень, коли виявляють системну лімфаденопатію чи абсолютний лімфоцитоз у загальному аналізі крові. У разі розвитку клінічних проявів хворі можуть скаржитися на загальне нездужання, швидку втомлюваність, анемічні симптоми (серцебиття, задишка, запаморочення, шум у вухах тощо), часті інфекційні захворювання вірусної чи бактеріальної природи. Значно рідше, ніж при лімфомі, у хворих на ХЛЛ виникають симптоми інтоксикації (B-): невмотивована гарячка, профузне пітніння, прогресуюча втрата маси тіла, частіше при прогресуванні чи трансформації захворювання. Найчастішими клінічними проявами ХЛЛ є системне збільшення, нерідко симетричне, периферичних лімфатичних вузлів, часом у вигляді конгломератів тістуватої консистенції. В окремих випадках відзначається збільшення селезінки та/чи печінки різного ступеня, гіпертрофія мигдаликів кільця Вальдеєра, іноді — збільшення інтраабдомінальних лімфовузлів. З прогресуванням захворювання в разі розвитку анемії відзначається блідість чи субіктеричність (при гемолізі) шкіри та слизової оболонки, іноді — геморагічні прояви як наслідок тромбоцитопенії (петехії, екхімози, кровоточивість слизової оболонки).
ДІАГНОСТИКА
Лабораторне обстеження
Обов’язкове:
- аналіз периферичної крові (визначення рівня гемоглобіну, еритроцитів, ретикулоцитів, кількості лейкоцитів, тромбоцитів, формула крові, тіні Гумпрехта, ШОЕ);
- стернальна пункція;
- біохімічний аналіз крові з визначенням рівня загального, прямого та непрямого білірубіну;
- визначення антиеритроцитарних та антитромбоцитарних антитіл;
- УЗД черевної порожнини з визначенням розмірів печінки та селезінки;
- рентгенографія органів грудної порожнини;
- імунофенотипування патологічних лімфоцитів периферичної крові та кісткового мозку.
Допоміжне:
- трепанобіопсія;
- біопсія, гістологічне дослідження лімфатичного вузла;
- визначення білкових фракцій;
- визначення рівня С-реактивного білка, лактатдегідрогенази, β2-мікроглобуліну;
- імуногістохімічне дослідження на зрізах лімфатичних вузлів;
- цитогенетичні дослідження;
- молекулярні дослідження.
Критерії встановлення діагнозу В-ХЛЛ:
- Постійний абсолютний лімфоцитоз периферичної крові, кількість лімфоцитів становить >5 Г/л, не пов’язане з іншими причинами.
- Мазок периферичної крові: переважають малі, морфологічно зрілі лімфоцити, визначаються тіні Гумпрехта.
- Імунофенотип патологічних лімфоцитів: СD5+ СD23+ СD20+dim sIg dim+.
Після встановлення діагнозу ХЛЛ визначається стадія захворювання за K.R. Rai (1975), J.L. Binet (1981) або згідно з рекомендаціями Міжнародної робочої наради з ХЛЛ (1989) (табл. 18.10–18.12).
Система стадіювання ХЛЛ за Rai (Rai K.R. et al., 1975)
Стадія | Клінічні характеристики |
0 | Лімфоцитоз у периферичній крові/кістковому мозку ізольовано |
I | Лімфоцитоз та лімфаденопатія |
II | Лімфоцитоз та гепатомегалія та/чи спленомегалія (+/– лімфаденопатія) |
III | Лімфоцитоз та анемія (гемоглобін <110 г/л) +/– лімфаденопатія, спленомегалія та/чи гепатомегалія |
IV | Лімфоцитоз і тромбоцитопенія (кількість тромбоцитів <100 Г/л) +/– анемія, лімфаденопатія, спленомегалія та/або гепатомегалія |
Система стадіювання ХЛЛ за Binet (1981)
Стадія | Клінічні характеристики |
А | Гемоглобін >100 г/л, тромбоцити >100 Г/л, залучення до процесу <3 лімфатичних зон |
В | Гемоглобін >100 г/л, тромбоцити >100 Г/л, залучення до процесу ≥3 лімфатичних зон |
С | Гемоглобін <100 г/л, або кількість тромбоцитів <100 Г/л, або поєднання цих ознак |
Стадії ХЛЛ згідно з рекомендаціями Міжнародної робочої наради з ХЛЛ (International Workshop on Chronic Lymphocytic Leukemia, 1989)
Стадії | Клінічні характеристики | Співвідношення за стадіями за Rai та Binet |
А | Відсутність анемії та тромбоцитопенії та залучення до процесу <3 лімфатичних зон | A(0), A(I), A(II) |
В | Відсутність анемії та тромбоцитопенії та залучення до процесу ≥3 лімфатичних зон | B(I), B(II) |
С | Анемія та/чи тромбоцитопенія незалежно від кількості уражених лімфатичних зон | C(III), C(IV) |
ПРОГНОСТИЧНІ ФАКТОРИ РИЗИКУ
Окрім cтадії захворювання, визнаними прогностичними чинниками ХЛЛ є так звані сироваткові маркери (рівень лактатдегідрогенази, β2-мікроглобуліну, тимідинкінази, розчиненого CD23), час подвоєння лімфоцитозу в периферичній крові, атипова морфологія лімфоцитів. Серед нових несприятливих прогностичних маркерів найбільш значущими є немутований статус важких ланцюгів IgVH, підвищена експресія протеїнкінази ZAP-70 у лейкемічних клітинах і CD38 на поверхні клітин, а також наявність цитогенетичних аномалій: del(17p), del(11q) та t(11q;v). Хромосомна аномалія del(13q) пов’язана зі сприятливим перебігом, тоді як прогностична значущість трисомії 12-ї хромосоми залишається суперечливою.
Важливим є питання початку терапії ХЛЛ. Темпи розвитку хвороби, швидкість збільшення кількості лейкоцитів, розміри лімфатичних вузлів та селезінки при ХЛЛ коливаються в широких межах. У терапії ХЛЛ панує принцип очікування, обґрунтований повільним доброякісним перебігом хвороби у частини пацієнтів. Пацієнт не потребує лікування доти, поки захворювання перебуває на стадії 0–I за Rai або A за Binet та відсутні ознаки прогресування захворювання. Доведено, що ранній початок терапії не збільшує тривалості життя хворих на ХЛЛ (Binet J.L. еt al., 1998). При розвитку інфекційних ускладнень проводиться симптоматична терапія. У разі прогресування захворювання спеціальне лікування слід розпочинати негайно (показання до початку терапії — табл. 18.13). Після закінчення всього обсягу лікування першої, другої або третьої лінії проводиться оцінка ефективності згідно з Критеріями відповіді на терапію за рекомендаціями Міжнародної робочої групи з ХЛЛ (2008) (табл. 18.14).
Показання до початку терапії
Прогресуючий лімфоцитоз: збільшення кількості лімфоцитів >50% за 2 міс, період подвоєння лімфоцитозу <6 міс |
Анемія (гемоглобін <110 г/л) та/чи тромбоцитопенія (кількість тромбоцитів <100 Г/л), що зумовлено інфільтрацією кісткового мозку |
Лихоманка без наявності інфекційного процесу (температура тіла ≥38 ˚С ≥2 тиж) |
Пітливість ≥1 міс |
Втрата маси тіла ≥10% за 6 міс |
Виражена загальна слабкість, ECOG ≥2 |
Аутоімунна анемія та/чи тромбоцитопенія |
Часті епізоди інфекційних ускладнень |
Масивна (>10 см) або прогресуюча лімфаденопатія |
Масивна (>6 см) або прогресуюча спленомегалія |
Гіперлейкоцитоз периферичної крові (кількість лейкоцитів периферичної крові >150 Г/л) |
Масивна інфільтрація кісткового мозку патологічними лімфоцитами (>80%) |
Критерії відповіді на терапію згідно з рекомендаціями Міжнародної робочої групи з ХЛЛ (International Workshop on Chronic Lymphocytic Leukemia, 2008)
Повна відповідь | Часткова відповідь | Прогресування | Стабілізація | |
Фізикальне обстеження (зокрема лімфовузлів, печінки, селезінки) | Норма | Зменшення на ≥50% | Збільшення на ≥ 50% чи поява нового пухлинного осередку | Зміни від –49% до +49% |
В-симптоми | Відсутні | |||
Циркулюючі клональні В-лімфоцити | Відсутні | Зменшення на ≥ 50% ініціального рівня | Збільшення на ≥ 50% ініціального рівня | Зміни від –49% до +49% |
Нейтрофільні гранулоцити (· 109/л) | >1,5 | >1,5 чи покращання на 50% порівняно з ініціальним рівнем | ||
Тромбоцити (· 109/л) | >100 | >100 чи покращання на 50% порівняно з ініціальним рівнем | Зниження на ≥50% порівняно з ініціальним рівнем (пов’язане з ХЛЛ) | Зміни від –49% до +49% |
Гемоглобін (г/л) | >110 (без гемотрансфузій та еритропоетину) | >110 чи покращання на 50% порівняно з ініціальним рівнем | Зниження >20 порівняно з ініціальним рівнем (пов’язане з ХЛЛ) | Підвищення <110 чи <50% порівняно з ініціальним рівнем чи зниження <20 |
Лімфоцити кісткового мозку (%) | <30 | ≥ 30% | Збільшення на ≥30% від норми | Немає змін порівняно з ініціальним рівнем |
Примітки | Тривалість>2 міс | Тривалість >2 міс | Синдром Ріхтера |
Лікування першої лінії
а) хворі віком <70 років*
- Флударабін + циклофосфамід + ритуксимаб (FCR).
- Флударабін + циклофосфамід (FC).
- Флударабін ± ритуксимаб.
*Флударабін може бути замінений на кладрибін. При застосуванні аналогів пурину слід передбачити профілактичне застосування ацикловіру (з метою профілактики герпесної інфекції) та ко-тримоксазолу (з метою профілактики пневмоцистної пневмонії).
б) хворі віком ≥70 років
- Хлорамбуцил ± преднізолон ± ритуксимаб.
- Циклофосфамід + вінкристин + преднізолон (COP).
- Флударабін ± ритуксимаб.
- Монотерапія флударабіном.
- Монотерапія метилпреднізолоном.
Терапія другої та наступних ліній (повторна терапія)
Попередня терапія може бути застосована повторно у хворих на ХЛЛ в разі розвитку рецидиву чи прогресування захворювання, щонайменше — через 1 рік. У разі більш раннього рецидивування, рефрактерності чи прогресії рекомендують такі режими лікування:
- Флударабін + циклофосфамід ± ритуксимаб — після хлорамбуцилу, схеми СОР чи іншого лікування без вмісту аналогів пурину (флударабін та ін.).
- СНОР ± ритуксимаб — у рефрактерних до аналогів пурину хворих чи рецидивах після флударабінвмісних режимів.
- Метилпреднізолон у високих дозах + ритуксимаб — при повторних рецидивах чи у рефрактерних до флударабін- та антрациклінвмісних режимів.*
*У разі наявності хромосомної аномалії del(17p) призначають метилпреднізолон у високих дозах.
